

Abstract
Gemcitabine, 2′-deoxy-2′, 2′-difluorocytidine (dFdC), is a drug approved for use against various solid tumors. Clinically, this moderately toxic nucleoside analog causes peripheral neuropathy, hematological dysfunction, and pulmonary toxicity in cancer patients. Although these side effects closely mimic symptoms of mitochondrial dysfunction, there is no direct evidence to show gemcitabine interferes with mitochondrial DNA replication catalyzed by human DNA polymerase γ. Here we employed presteady state kinetic methods to directly investigate the incorporation of the 5′-triphosphorylated form of gemcitabine (dFdCTP), the excision of the incorporated monophosphorylated form (dFdCMP), and the bypass of template base dFdC catalyzed by human DNA polymerase γ. Opposite template base dG, dFdCTP was incorporated with a 432-fold lower efficiency than dCTP. Although dFdC is not a chain terminator, the incorporated dFdCMP decreased the incorporation efficiency of the next 2 correct nucleotides by 214- and 7-fold, respectively. Moreover, the primer 3′-dFdCMP was excised with a 50-fold slower rate than the matched 3′-dCMP. When dFdC was encountered as a template base, DNA polymerase γ paused at the lesion and one downstream position but eventually elongated the primer to full-length product. These pauses were because of a 1,000-fold decrease in nucleotide incorporation efficiency. Interestingly, the polymerase fidelity at these pause sites decreased by 2 orders of magnitude. Thus, our pre-steady state kinetic studies provide direct evidence demonstrating the inhibitory effect of gemcitabine on the activity of human mitochondrial DNA polymerase.
Many nucleoside analogs are potent anti-cancer and antiviral drug compounds. Among 15 Food and Drug Administration-approved nucleoside analogs, gemcitabine or 2′-deoxy-2′,2′-difluorocytidine (dFdC,4 supplemental Fig. 1) is an anti-cancer drug that is clinically used for the treatment of non-small cell lung cancer (1), pancreatic cancer (2), metastatic breast cancer (3), and ovarian cancer (4). It has also shown promising efficacy for the treatment of other solid tumors and hematological malignancies (5-12) suggesting more wide-spread use in the future. In addition to its use as a monotherapy, gemcitabine is often most effective when used as part of a combination therapy, frequently with platinum-based and topoisomerase-targeted chemotherapeutic agents (13-15).
Gemcitabine is administered in the form of a biologically inactive prodrug that first permeates the cellular membrane by facilitated diffusion (16, 17) almost exclusively via the human equilibrative nucleoside transporter number 1 (17, 18). Following transport, dFdC is metabolized to the biologically active monophosphorylated form (dFdCMP) by deoxycytidine kinase, which is the rate-limiting step during the activation of gemcitabine (19). Subsequently, dFdCMP is further phosphorylated to form the cytotoxic metabolites gemcitabine diphosphate (dFdCDP) and gemcitabine triphosphate (dFdCTP) by cellular kinases. It has been shown that dFdCTP competes effectively against endogenous dCTP for incorporation into genomic DNA (20) and against CTP into RNA (21), and that the proof-reading exonuclease activity of human DNA polymerase ε is essentially unable to remove dFdCMP once incorporated into DNA (22). Interestingly, dFdCTP incorporation by human DNA polymerase α results in “masked termination” of DNA synthesis where, following a single dFdCTP incorporation into DNA, the primer is extended by only one additional dNTP before polymerization is inhibited (20, 22). However, in addition to being incorporated into DNA and RNA, dFdCDP and dFdCTP are known to inhibit ribonucleotide reductase, thereby significantly decreasing cellular dCTP concentrations and leading to increased phosphorylation of dFdCDP (23, 24). Furthermore, high concentrations of dFdCTP inhibit CTP synthetase, whereby dCTP and CTP pools are further decreased (25, 26). Reduced competition from diminished dCTP pools makes dFdCTP incorporation into DNA and RNA more probable, thus promoting cell cycle arrest and apoptosis and inhibiting DNA repair (23). Furthermore, dFdCMP and dFdCTP also inhibit dCMP deaminase, the major pathway by which dFdCMP is metabolized (27). The combined synergistic effect of these inhibitory activities is termed “self-potentiation” and is illustrated in supplemental Fig. 2 (28, 29).
Moderate toxicity of gemcitabine has been observed in cancer patients with peripheral neuropathy (30, 31) and hematological dysfunction in which myelosuppression frequently emerges as the dose-limiting factor (4). The toxicological profile of gemcitabine resembles that of many other anti-viral nucleoside analogs and frequently mimics the symptoms of heritable mitochondrial defects (32). Furthermore, the unique susceptibility of the mitochondria to the toxic effects of nucleoside analog drugs makes it a prime suspect as the cause of gemcitabine-induced toxicity. An excellent precedent to support this hypothesis is the case of a related nucleoside analog drug developed by Lilly in the early 1990s. Fialuridine (FIAU), a nucleoside analog drug similar to gemcitabine but targeted toward hepatitis B, killed five patients in clinical trials (33, 34) due to the fact that FIAU is an excellent substrate for human mitochondrial DNA polymerase holoenzyme (pol γ) (33). Previously, we have used pre-steady state kinetic methods to evaluate the mitochondrial toxicity of several anti-HIV nucleoside analogs and the anti-hepatitis B nucleoside analog FIAU with recombinant pol γ (35). Our in vitro kinetic data correlate well with the observed toxicities of these drugs in vivo (36). Therefore, to evaluate the potential mitochondrial toxicity of gemcitabine, we again employed pre-steady state kinetic methods to evaluate the incorporation, extension, and excision of gemcitabine catalyzed by pol γ. In addition, we examined whether or not an incorporated gemcitabine as a template base might lead to mutations in the next round of mitochondrial DNA synthesis. Our resulting data provide direct evidence for the inhibitory effect of gemcitabine on the activity of human pol γ.
EXPERIMENTAL PROCEDURES
Materials—What follows is a list of the reagents used for these experiments and their sources: [γ-32P]ATP from GE Healthcare; Bio-Spin columns from Bio-Rad; dNTPs from Invitrogen; dFdCTP from Trilink Biotechnologies Inc. (San Diego); OptiKinase from U. S. Biochemical Corp.
Optimized Reaction Buffer G—Buffer G consisted of 50 mm Tris-Cl, pH 7.5, at 37 °C, 100 mm NaCl, and 2.5 mm MgCl2. Note that all concentrations listed in this paper refer to the final concentration after mixing unless otherwise noted.
Optimized Reaction Buffer L—Buffer L consisted of 50 mm Tris-Cl, pH 8.4, at 37 °C, 100 mm NaCl, 5 mm MgCl2, 0.1 mm EDTA, 5 mm dithiothreitol, 0.1 mg/ml bovine serum albumin, and 10% glycerol.
Optimized Reaction Buffer M—Buffer M consisted of 50 mm HEPES, pH 8.0, at 25 °C, 12 mm NaCl, 8.75 mm MgCl2, 0.2 mm EDTA, 5 mm dithiothreitol, 0.1 mg/ml bovine serum albumin, and 10% glycerol.
Purification of Human Polymerase γ Subunits—Expression and purification of wild-type human DNA polymerase γ, its exonuclease-deficient mutant E200A, and the small accessory subunit were carried out as described previously (35, 37).
Synthetic Oligodeoxyribonucleotides—All DNA substrates not containing gemcitabine were purchased from Integrated DNA Technologies (Coralville, IA) and purified by denaturing PAGE (17% acrylamide, 8 m urea). Concentrations of synthetic oligodeoxyribonucleotides were determined from their UV absorbance at 260 nm. Primers were 5′-32P-labeled by incubation with [γ-32P]ATP and OptiKinase at 37 °C for 1 h. The remaining [γ-32P]ATP was subsequently removed by size exclusion chromatography in a Bio-Spin 6 column. All primers were annealed to their respective templates in a 1:1.15 (primer: template) molar ratio by heating the mixture to 95 °C for 10 min and then slowly cooling to room temperature over ∼6 h.
Synthetic Oligodeoxyribonucleotides Containing Gemcitabine—To create two DNA primers and a template that contain site-specific gemcitabine, a primer extension and ligation strategy was employed. Primer 23F-mer (Table 1) was synthesized by mixing DNA 22/41-mer (Table 1) with 5 μm dFdCTP and human DNA polymerase μ (38), a template-directed DNA polymerase capable of efficiently incorporating dFdCTP, in reaction buffer M. The reaction was conducted for 2 h at 25 °C yielding maximum conversion of the 22-mer primer to 23F-mer (Table 1). A simultaneous control reaction was performed under identical conditions but substituting dCTP for dFdCTP. The products of both reactions were radiolabeled and compared using sequencing PAGE (17% acrylamide, 8 m urea) at single nucleotide resolution. The position of the 3′-dFdCMP-terminated 23F-mer was determined by comparison with the control reaction. The 23F-mer product was then gel-purified, thereby simultaneously removing any remaining unreacted 22-mer primer and any multiple incorporation products. A similar reaction and purification scheme was performed to synthesize primer 24FG-mer (Table 1) except 23F-mer was used as the starting primer.
TABLE 1.
DNA substrates
| 15/41-mer | 5′-GGACGGCATTGGATC |
| 3′-CCTGCCGTAACCTAGCTGCCACTCAACCAACCTGCCGACGC-5′ | |
| 22/41-mer | 5′-CGCAGCCGTCCAACCAACTCAC |
| 3′-GCGTCGGCAGGTTGGTTGAGTGGCAGCTAGGTTACGGCAGG-5′ | |
| 23/41-mer | 5′-CGCAGCCGTCCAACCAACTCACC |
| 3′-GCGTCGGCAGGTTGGTTGAGTGGCAGCTAGGTTACGGCAGG-5′ | |
| 23F/41-mera | 5′-CGCAGCCGTCCAACCAACTCACF |
| 3′-GCGTCGGCAGGTTGGTTGAGTGGCAGCTAGGTTACGGCAGG-5′ | |
| 24FG/41-mera | 5′-CGCAGCCGTCCAACCAACTCACFG |
| 3′-GCGTCGGCAGGTTGGTTGAGTGGCAGCTAGGTTACGGCAGG-5′ | |
| 25FGT/41-mera | 5′-CGCAGCCGTCCAACCAACTCACFGT |
| 3′-GCGTCGGCAGGTTGGTTGAGTGGCAGCTAGGTTACGGCAGG-5′ | |
| 15/41F-mera | 5′-GGACGGCATTGGATC |
| 3′-CCTGCCGTAACCTAGCTGCFACTCAACCAACCTGCCGACGC-5′ | |
| 18/41F-mera | 5′-GGACGGCATTGGATCGAC |
| 3′-CCTGCCGTAACCTAGCTGCFACTCAACCAACCTGCCGACGC-5′ | |
| 19/41F-mera | 5′-GGACGGCATTGGATCGACG |
| 3′-CCTGCCGTAACCTAGCTGCFACTCAACCAACCTGCCGACGC-5′ | |
| 20/41F-mera | 5′-GGACGGCATTGGATCGACGG |
| 3′-CCTGCCGTAACCTAGCTGCFACTCAACCAACCTGCCGACGC-5′ | |
| 20T/41F-mera | 5′-GGACGGCATTGGATCGACGT |
| 3′-CCTGCCGTAACCTAGCTGCFACTCAACCAACCTGCCGACGC-5′ | |
| 21/41F-mera | 5′-GGACGGCATTGGATCGACGGT |
| 3′-CCTGCCGTAACCTAGCTGCFACTCAACCAACCTGCCGACGC-5′ | |
| 22/41F-mera | 5′-GGACGGCATTGGATCGACGGTG |
| 3′-CCTGCCGTAACCTAGCTGCFACTCAACCAACCTGCCGACGC-5′ | |
| 25/45-merb | 5′-GCCTCGCAGCCGTCCAACCAACTCA |
| 3′-CGGAGCGTCGGCAGGTTGGTTGAGTTGGAGCTAGGTTACGGCAGG-5′ | |
| 21-19/35-merc | 5′-CGCAGCCGTCCAACCAACTCACG TCGATCCAATGCCGTCC-3′ |
| 3′-GCGTCGGCAGGTTGGTTGAGTGGCAGCTAGGTTAC-5′ | |
| 21F-19/35-merc | 5′-CGCAGCCGTCCAACCAACTCAF CGTCGATCCAATGCCGTCC-3′ |
| 3′-GCGTCGGCAGGTTGGTTGAGTG-GCAGCTAGGTTAC-5′ |
F denotes gemcitabine.
The template base opposite primer at the 26th base was varied to allow correct base pairing of incoming dNTP.
The downstream strand 19-mer of substrates 21-19/35-mer and 21F-19/35-mer were 5′-phosphorylated.
To synthesize template 41F-mer (Table 1), the DNA substrate 21-19/35-mer (Table 1) was incubated with 8 μm dFdCTP and human DNA polymerase λ (39), a template-directed, gap-filling DNA polymerase capable of efficiently incorporating dFdCTP, for 5 min at 37 °C in reaction buffer L to form the 21F-19/35-mer. Unreacted dFdCTP was then removed using gel filtration (Bio-Spin 6, Bio-Rad). The DNA solution was heated to 95 °C for 10 min and then slowly cooled to room temperature over 6 h to re-anneal 21F-19/35-mer (a nicked DNA substrate). A solution of 10 mm MgCl2, 1 mm ATP, and T4 DNA ligase (11 units/μl) was added to the annealed DNA solution to ligate the nicked DNA for 7 min at 37 °C. The resulting 41F-mer was radiolabeled and compared with the control DNA template 41-mer (Table 1) using denaturing PAGE at single nucleotide resolution, thereby verifying both the incorporation of dFdCMP and the subsequent ligation of the primers. The resulting 41F-mer was then gel-purified to ensure homogeneity. Furthermore, the incorporation and position of the dFdCMP moiety into the 41F-mer template was additionally verified through observation of the pause pattern exhibited by pol γ as it encountered the dFdCMP moiety at the expected position in the template.
Single-turnover Nucleotide Incorporation Assay—All assays using pol γ were carried out at 37 °C in buffer G containing 2.5 mm MgCl2. For single nucleotide incorporation assays, pol γ (90 nm) and its cofactor SSU (450 nm) were combined (1:5 molar ratio) and preincubated on ice in buffer G for 20 min to form human pol γ holoenzyme. Next, 30 nm of a DNA substrate containing a 5′-[32P]-labeled DNA primer was added to the reconstituted holoenzyme (3:1 molar ratio, holoenzyme:DNA) and incubated on ice for an additional 20 min. The single nucleotide incorporation reaction was initiated by the addition of dNTP and 2.5 mm MgCl2 in buffer G using a rapid chemical quench apparatus (KinTek, Clarence, PA). After varying reaction times at 37 °C, the reactions were quenched by the addition of 0.37 m EDTA.
Excision Reactions—For the 3′ → 5′-exonuclease assay, wild-type pol γ (100 nm) and SSU (500 nm) in buffer G were first preincubated on ice for 20 min to form human pol γ holoenzyme and then mixed with 5′-32P-labeled DNA substrate (75 nm) in the absence of Mg2+. The 3′ → 5′-exonuclease reaction was initiated by the addition of 2.5 mm MgCl2 in buffer G using a rapid chemical quench apparatus. After varying reaction times at 37 °C, the reactions were quenched by the addition of 0.37 m EDTA. The concentration of remaining full-length primer as a function of time was quantitated, and the exonuclease reaction time course (Fig. 2) was fit to Equation 3 or 4 to yield an excision rate constant.
FIGURE 2.

Measurement of the rate constant of DNA primer degradation by the 3′ → 5′-exonuclease proofreading activity of the wild-type pol γ. A preincubated solution of the wild-type pol γ (100 nm), pol γ accessory subunit (500 nm), and 23/41-mer or 23F/41-mer (75 nm) in buffer G was rapidly mixed with 2.5 mm MgCl2 and reacted for various time intervals. The excision reaction was quenched by the addition of 0.37 m EDTA. The concentration of the remaining full-length primer versus time was plotted and fit to Equation 3 (23/41-mer, ▪; 23F/41-mer, •) yielding a kexo of 0.06 ± 0.02 s-1 for 23/41-mer and 0.0011 ± 0.0001 s-1 for 23F/41-mer.
Running Start Nucleotide Incorporation Assay—For the running start nucleotide incorporation assay, a DNA substrate (30 nm) was first preincubated with a solution of pol γ (90 nm) and SSU (450 nm) in buffer G as described above. This solution was rapidly mixed with MgCl2 (2.5 mm) and dNTPs (100 μm each). The primer elongation at various times was stopped by the addition of 0.37 m EDTA.
Product Analysis—Products of the polymerase and exonuclease reactions were separated by sequencing gel electrophoresis (17% acrylamide, 8 m urea, 1× TBE running buffer) and quantitated using a PhosphorImager 445 SI (GE Healthcare).
Data Analysis—Kinetic data were fit via nonlinear regression using KaleidaGraph (Synergy Software). Data from single-turnover nucleotide incorporation assays were fit to a single exponential (Equation 1) to obtain an observed incorporation rate constant (kobs). The dNTP concentration dependence of kobs was fit to a hyperbolic equation (Equation 2) to yield both the equilibrium dissociation constant (Kd) and the maximum nucleotide incorporation rate constant (kp). Single-phase exonuclease reaction time courses were fit to a single exponential equation (Equation 3) to yield the exonuclease rate constant (kexo). Biphasic exonuclease reaction time courses were fit to a double exponential (Equation 4) to yield kexo,1 and reaction amplitude A1 in the fast phase and kexo,2 and reaction amplitude A2 in the slow phase.
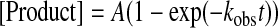 |
(Eq.1) |
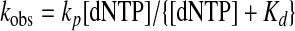 |
(Eq.2) |
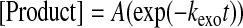 |
(Eq.3) |
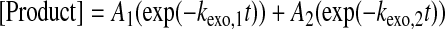 |
(Eq.4) |
RESULTS
Determination of the Pre-steady State Kinetic Parameters for dFdCTP and dCTP Incorporation—The kinetic mechanism of DNA polymerization catalyzed by human pol γ holoenzyme has been established by using pre-steady state kinetic analysis (35, 37, 40, 41). This mechanism shows that an incoming dNTP binds to the pol γ·DNA binary complex to establish a rapid equilibrium prior to nucleotide incorporation (37, 41). Therefore, the ground state equilibrium dissociation constant of an incoming dNTP (Kd) and its maximum incorporation rate constant (kp) can be measured by observing the nucleotide concentration dependence of the observed single-turnover rate constant (kobs) (40). To examine the toxicity of gemcitabine toward human mitochondria resulting from inhibition of pol γ, we first determined the substrate specificity (kp/Kd) of dFdCTP. Because a nucleotide analog is usually incorporated slowly, and its incorporation rate constant is comparable or smaller than the dissociation rate constant of DNA from the enzyme·DNA binary complex, the burst phase is either insignificant or does not exist. Thus, the experiments to measure the kp/Kd value of dFdCTP were performed with pol γ in molar excess over DNA to allow the direct observation of nucleotide incorporation in a single pass of the reactants through the catalytic cycle without complications resulting from the steady state formation of products (42). In addition, because the wild-type pol γ has highly efficient 3′ → 5′-exonuclease activity (41), which excises a primer from its 3′ terminus and thereby complicates direct observation of the incorporation of dFdCTP, we used pol γ E200A, a well characterized single point mutant at the 3′ → 5′-exonuclease active site, to determine the kinetics of dFdCTP incorporation (40). This mutant incorporates normal nucleotides with similar kinetics as the wild-type pol γ but is exonuclease-deficient (36).
To measure the pre-steady state kinetic parameters for dFdCTP incorporation, a preincubated solution of pol γ E200A (90 nm large subunit, 450 nm accessory subunit) and 5′-32P-labeled 22/41-mer (30 nm, Table 1) was mixed and reacted with increasing concentrations of dFdCTP at 37 °C for various times. An autoradiograph gel image (Fig. 1A) showed that E200A gradually incorporated dFdCTP, as primer 22-mer was elongated to 23-mer. Each time course of product formation in Fig. 1B was fit to Equation 1 (see “Experimental Procedures”) to yield an observed single-turnover rate constant, kobs. The kobs values were then plotted as a function of dFdCTP concentration (Fig. 1C). The data were fit to Equation 2 (see “Experimental Procedures”) to yield a kp of 2.0 ± 0.3 s-1 and a Kd of 21 ± 7 μm (Table 2). The substrate specificity of dFdCTP was calculated to be 0.095 μm-1 s-1. Similarly, we measured the kp (37 ± 2 s-1) and Kd (0.9 ± 0.2 μm) for the incorporation of dCTP into the 22/41-mer (Table 1) under single-turnover reaction conditions (data not shown). These kinetic parameters agreed well with those that were measured under burst reaction conditions (40), thus validating our single-turnover approach. The incorporation efficiency of dCTP was then calculated to be 41 μm-1 s-1. Thus, the discrimination, defined as the efficiency ratio of (kp/Kd)dCTP/(kp/Kd)dFdCTP, exhibited by the polymerase activity of human pol γ against dFdCTP was 432-fold (Table 2).
FIGURE 1.

Concentration dependence of observed single-turnover rate constant of dFdCTP incorporation. A, autoradiograph gel image shows the incorporation of dFdCTP (16 μm) catalyzed by a pol γ mutant E200A. B, preincubated solution of E200A (90 nm), pol γ accessory subunit (450 nm), and 5′-32P-labeled 22/41-mer (30 nm) was rapidly mixed with increasing concentrations of dFdCTP·Mg2+ (2.4 μm, •; 4.9 μm, ○; 9.7 μm, ▪; 19.5 μm, □;39 μm, ▴; 78 μm, ▵) and reacted at 37 °C for increasing times. The solid lines were fit to Equation 1 using nonlinear regression which determined the observed rate constants, kobs. C, kobs values were plotted as a function of dFdCTP concentration. The data (•) were fit to Equation 2 using nonlinear regression, thus yielding a kp of 2.0 ± 0.3 s-1 and a Kd of 21 ± 7 μm.
TABLE 2.
Kinetic parameters of single nucleotide incorporation catalyzed by pol γ E200A under single-turnover conditions at 37 °C
Measurement of the Excision Rate Constants of Matched 3′-dFdCMP and 3′-dCMP—The 3′ → 5′-exonuclease activity of human pol γ recognizes mismatched 3′-base(s) in a DNA primer and rapidly excises 1-7 mismatched bases at a rate of 1-9 nucleotides/s but slowly excises the matched 3′-terminal base of a primer (41). Although paired with template base dG, the incorporated 3′-dFdCMP in a primer may be a substrate for this 3′ → 5′ exonucleolytic proofreading mechanism and thereby excised. We first synthesized and purified primer 23F-mer (see “Experimental Procedures” and Table 1) and then measured the excision rate constant (kexo) of 3′-dFdCMP from the DNA substrate 23F/41-mer (Table 1) by the wild-type pol γ under single-turnover reaction conditions. A preincubated solution of the wild-type pol γ (100 nm large subunit, 500 nm accessory subunit) and 75 nm 5′-[32P]DNA (23/41-mer or 23F/41-mer) in buffer G was rapidly mixed and reacted with 2.5 mm MgCl2 for various time intervals prior to being quenched with 0.37 m EDTA. The concentration of the remaining full-length primer versus reaction time was plotted and fit to Equation 3 (see “Experimental Procedures”), yielding the kexo of 0.06 ± 0.02 and 0.0011 ± 0.0001 s-1 for the 23/41-mer and 23F/41-mer, respectively (Fig. 2 and Table 3). The kexo of 23/41-mer DNA was similar to 0.05 ± 0.01 s-1 measured previously with a completely matched substrate 25/45-mer (Table 1) (41). Interestingly, the excision of the matched 3′-dFdCMP moiety by the wild-type pol γ was 55-fold slower than the excision of matched 3′-dCMP. This suggested that an incorporated dFdCMP likely escaped the editing process and subsequently would be embedded into the mtDNA.
TABLE 3.
Excision rate constants for the 3′ → 5′ -exonuclease activity of the wild-type human pol γ holoenzyme under single-turnover conditions at 37 °C
| DNA | Fast phase kexo, 1 | Fast phase amplitude | Slow phase kexo, 2 | Slow phase amplitude |
|---|---|---|---|---|
| s−1 | % | s−1 | % | |
| 23/41-mer | 0.06 ± 0.02 | NAa | NA | NA |
| 23F/41-mer | 0.0011 ± 0.0001 | NA | NA | NA |
| 20/41F-mer | 0.028 ± 0.006 | NA | NA | NA |
| 20T/41F-mer | 0.2 ± 0.2 | 17 ± 8 | 0.008 ± 0.002 | 83 ± 8 |
NA means not applicable.
Measurement of the Extension Efficiency of a Primer Terminated with 3′-dFdCMP—It is possible that an incorporated dFdCMP moiety on the 3′ terminus of a DNA primer could significantly alter the ability of pol γ to extend that primer. To examine this possibility, we measured the kinetic parameters for the incorporation of correct dGTP into 23F/41-mer (Table 1) catalyzed by E200A under single-turnover conditions as described above (data not shown). dGTP was incorporated with a kp of 1.5 ± 0.1 s-1, a Kd of 7.2 ± 0.9 μm, and a kp/Kd of 0.21 μm-1 s-1 (Table 2). In comparison, a matched dGTP is incorporated into normal 25/45-mer (Table 1) with a kp of 37 ± 2 s-1 and a Kd of 0.8 ± 0.1 μm, which defined a kp/Kd of 45 μm-1 s-1 (40). Thus, the 3′-dFdCMP decreased the incorporation efficiency of the first downstream nucleotide by 214-fold, suggesting that an incorporated gemcitabine was problematic for primer extension. Such an inhibitory effect may persist beyond 1 nucleotide. Therefore, to examine if an embedded gemcitabine inhibited the incorporation of the second downstream nucleotide, we prepared primer 24FG-mer (see “Experimental Procedures”). We then measured the kinetic parameters of correct dTTP incorporation into 24FG/41-mer (Table 1) catalyzed by E200A (data not shown). Under single-turnover reaction conditions as described above, dTTP was incorporated with a kp of 2.8 ± 0.1 s-1, a Kd of 0.5 ± 0.1 μm, and a kp/Kd of 5.6 μm-1 s-1 (Table 2). In comparison, given a canonical DNA substrate of 25/45-mer (Table 1), correct dTTP is incorporated with a kp of 25 s-1, a Kd of 0.6 μm, and a kp/Kd of 39 μm-1 s-1 (Table 4) (40). Thus, primer 3′-dFdCMP lowered the second downstream nucleotide incorporation efficiency by 7-fold. Interestingly, the third nucleotide (dCTP) downstream from the embedded dFdCMP was incorporated into 25FGT/41-mer with similar efficiency as it was incorporated into normal 22/41-mer (data not shown). Therefore, an incorporated dFdCMP in the DNA primer only affected two downstream nucleotide incorporation events, especially the first one. However, it did not terminate primer elongation.
TABLE 4.
Kinetic parameters of single nucleotide incorporation into DNA containing a template base dFdCMP catalyzed by pol γ E200A under single-turnover conditions at 37 °C
| DNA | dNTP | Kd | kp | kp/Kd | Efficiency ratioa |
|---|---|---|---|---|---|
| μM | s−1 | μM−1s−1 | |||
| 18/41F-mer | dGTP | 1.1 ± 0.2 | 2.1 ± 0.1 | 1.9 | 2.4 × 101 |
| 19/41F-mer | dGTP | 150 ± 10 | 6.3 ± 0.2 | 0.042 | 1.1 × 103 |
| 20/41F-mer | dTTP | 6.0 ± 0.5 | 0.11 ± 0.01 | 0.018 | 2.2 × 103 |
| 21/41F-mer | dGTP | 5 ± 2 | 1.7 ± 0.4 | 0.34 | 1.3 × 102 |
| 22/41F-mer | dATP | 0.7 ± 0.1 | 8.3 ± 0.3 | 12 | 4.8 |
| 25/45-merb | dATP | 0.8 ± 0.1 | 45 ± 1 | 57 | |
| 25/45-merb | dTTP | 0.6 ± 0.2 | 25 ± 2 | 39 | |
| 25/45-merb | dGTP | 0.8 ± 0.1 | 37 ± 2 | 45 | |
| 25/45-merb | dCTP | 0.9 ± 0.2 | 43 ± 2 | 47 |
Data are calculated as (kp/Kd)correct dNTP into 25/45-mer/ (kp/Kd)dNTP into DNA containing a template base dFdCMP.
Kinetic parameters are from Ref. 40.
Running Start Primer Extension Assays—To investigate whether or not a dFdC lesion embedded in a DNA template affects DNA synthesis catalyzed by human pol γ, we first synthesized template 41F-mer (Table 1) as described under “Experimental Procedures.” To examine the nucleotide incorporation profile, a running start primer elongation assay was performed to evaluate the ability of the wild-type pol γ (90 nm) to bypass the template dFdCMP lesion in 15/41F-mer (30 nm, Table 1) in the presence of four dNTPs (100 μm each). In comparison, a similar running start assay was also performed with an undamaged control substrate 15/41-mer (30 nm, Table 1). With the 15/41-mer, the wild-type pol γ holoenzyme was able to extend the 15-mer primer to the full-length 41-mer in 1 s (Fig. 3A). This rate was consistent with the average maximum rate constant (38 s-1) of single nucleotide incorporation into normal 25/45-mer (Table 4) (40). In contrast, the wild-type pol γ paused opposite the dFdCMP lesion and 1 base downstream of the dFdCMP lesion in the 15/41F-mer (Fig. 3B). Consequently, the full-length product 41-mer was formed in 3 s, rather than just 1 s as observed with control 15/41-mer (Fig. 3A). Although the synthesis of the 41-mer was delayed, Fig. 3B showed that the wild-type pol γ eventually bypassed the dFdCMP lesion.
FIGURE 3.

Running start primer elongation catalyzed by the wild-type pol γ and the exonuclease-deficient mutant E200A. A solution of control substrate 15/41-mer (A and C, 30 nm each) or DNA substrate 15/41F-mer (B and D, 30 nm each), preincubated with the wild-type pol γ (A and B, 90 nm) or E200A (C and D, 90 nm) and pol γ accessory subunit (450 nm) in buffer G, was rapidly mixed with 2.5 mm MgCl2 and 100 μm each of dATP, dCTP, dGTP, and dTTP. Reactions were allowed to continue for various time intervals before being quenched by the addition of 0.37 m EDTA. Product lengths and the position of the embedded dFdC moiety in the template are indicated.
To examine whether or not the 3′ → 5′-exonuclease activity of human pol γ plays any role in the bypass of the dFdCMP lesion, we performed the same running start primer elongation assays with pol γ E200A. Fig. 3, C and D, showed that the product formation patterns with both 15/41-mer and 15/41F-mer were almost identical to the patterns in Fig. 3, A and B, respectively. Thus, the 3′ → 5′-exonuclease activity appeared not to recognize template base dFdC as a lesion and was dispensable for its bypass.
Measurement of the Excision Rate Constant of Primer 3′-dNMP Opposite Template Base dFdCMP—To quantitatively interrogate the effect of an embedded dFdCMP moiety on the 3′ → 5′-exonuclease activity of pol γ, we synthesized two DNA substrates (see “Experimental Procedures”) as follows: 20/41F-mer and 20T/41F-mer (Table 1) which contain a 3′-terminal “correct” base pair and a mispair, respectively. Under single-turnover conditions, the wild-type pol γ holoenzyme was found to excise the primer 3′-bases of the 20/41F-mer with a kexo of 0.028 ± 0.006 s-1 (Table 3). This kexo value was roughly 2-fold slower than the kexo of 0.06 ± 0.02 s-1 observed with a normal substrate 23/41-mer (Table 3). In comparison, the time course of the cleavage of the 20T/41F-mer by the wild-type pol γ under single-turnover reaction conditions was biphasic and fit to Equation 4 (see “Experimental Procedures”) to yield a kexo,1 of 0.2 ± 0.2 s-1 and an amplitude of (17 ± 8)% in the fast phase and a kexo,2 of 0.008 ± 0.002 s-1 and an amplitude of (83 ± 8)% in the slow phase (data not shown). Similar biphasic kinetics are also observed previously with the cleavage of normal DNA containing a single 3′-mismatched base but with larger kexo values in the fast phase (1.1 s-1) and slow phase (0.04 s-1) (40). These data confirmed that template base dFdC inhibited the proofreading activity of human pol γ.
Measurement of Incorporation Efficiency of Nucleotides Opposite Template dFdCMP—The reason that pol γ strongly paused in Fig. 3, B and D, is likely because the dFdCMP lesion altered the local template structure and significantly decreased the incorporation efficiencies of adjacent nucleotides. To evaluate this hypothesis, we measured the incorporation efficiencies of correct nucleotides into 18/41F-mer, 19/41F-mer, 20/41F-mer, 21/41F-mer, and 22/41F-mer (Table 1) catalyzed by pol γ E200A under single-turnover reaction conditions (data not shown). The measured kinetic parameters in Table 4 indicated that, relative to the corresponding values with normal 25/45-mer (Table 1) (40), the ground-state binding affinity (1/Kd) of a correct incoming nucleotide was up to 2 orders of magnitude lower at the two strong pause sites but was within 5-fold at the non-pause sites. In comparison, the maximum nucleotide incorporation rate constant dropped by 3 orders of magnitude at the second strong pause site, whereas the kp values at other positions were 5-30-fold lower than the corresponding parameters given for 25/45-mer (Table 4). As expected, the substrate specificity (kp/Kd) values were more informative in demonstrating why pol γ paused at the two positions in Fig. 3, B and D. Before encountering the template base dFdCMP, pol γ E200A incorporated correct dGTP into 18/41F-mer with only 24-fold lower substrate specificity compared with normal 25/45-mer (Table 4). The efficiency ratio increased from 2.4 × 10-1 to 1.1 × 103 when pol γ E200A incorporated dGTP opposite dFdCMP and to 2.2 × 103 when pol γ E200A incorporated dTTP into 20/41F-mer to extend the primer and bypass dFdCMP. These significant decreases in the efficiency ratio indicated that pol γ was inefficient at incorporating nucleotides at these two positions and paused (Fig. 3, B and D). After pol γ bypassed the dFdCMP, the efficiency ratios for the next two downstream nucleotide incorporations (Table 4) decreased to 130 and 4.8, respectively. Although we did not measure the substrate specificity of dGTP incorporation into 23/41F-mer, we expect the inhibitory effect of an embedded gemcitabine in the template will disappear at this position and further downstream. Notably, based on the low nucleotide incorporation efficiency with 21/41F-mer (Table 4), pol γ E200A was expected to pause at this position. However, the pause was not obvious in Fig. 3D. This discrepancy was because the kp of 1.7 s-1 is relatively fast, and the reaction times were relatively long.
Measurement of Nucleotide Incorporation Fidelity at the Pause Sites—Opposite dFdCMP, pol γ may favor mismatched dNTPs over dGTP. To examine this possibility, each dNTP (200 μm) was reacted individually with a preincubated solution of E200A and 19/41F-mer (Table 1) for 15 s or 1 h. The supplemental Fig. 3A showed that pol γ E200A preferred to incorporate dGTP opposite dFdCMP at both time intervals. Interestingly, dTTP was mis-incorporated multiple times. This suggested that pol γ E200A may have a high tendency to misincorporate dTTP. To check if this was the case, we measured the kp/Kd value for dTTP incorporation into 19/41F-mer under single-turnover conditions (data not shown). The calculated fidelity in Table 5 indicated that pol γ E200A only favored dGTP over dTTP by 3,000-fold, which was much lower than the 6.4 × 105-fold observed with normal 25/45-mer (43). Thus, the fidelity of nucleotide incorporation opposite dFdCMP was lowered by 213-fold.
TABLE 5.
Fidelity at the two strong pause sites
| DNA | dNTP | Kd | kp | kp/Kd | Fidelitya |
|---|---|---|---|---|---|
| μm | s−1 | μm−1 s−1 | |||
| 19/41 F-mer | dGTP | 150 ± 10 | 6.3 ± 0.2 | 4.2 × 10−2 | 1 |
| 19/41 F-mer | dTTPb | 110 ± 30 | 0.0015 ± 0.0002 | 1.4 × 10−5 | 3.0 × 103 |
| 20/41 F-mer | dTTP | 6.0 ± 0.5 | 0.11 ± 0.01 | 1.8 × 10−2 | 1 |
| 20/41 F-mer | dATPb | 120 ± 20 | 0.0059 ± 0.0003 | 4.9 × 10−5 | 3.7 × 102 |
Data are calculated as (kp/Kd) correct dNTP / (kp/Kd) incorrect dNTP.
Boldface type indicates incorrect nucleotide.
To further examine the effect of template lesion dFdCMP on polymerase fidelity, we tested whether or not pol γ was errorprone when extending primer 20-mer to 21-mer. Opposite template base dAMP, pol γ E200A incorporated correct dTTP more efficiently than incorrect nucleotides (supplemental Fig. 3B), and dATP was the most favored incorrect nucleotide. We further measured the kinetic parameters of dATP misincorporation into 20/41F-mer under single-turnover conditions (data not shown). pol γ E200A favored correct dTTP over incorrect dATP by only 370-fold (Table 5), which was 757-fold lower than the corresponding fidelity (2.8 × 105) observed with canonical 25/45-mer (43).
DISCUSSION
Konerding et al. (44) have solved the solution-phase structure of an Okazaki fragment (12-bp) with an internally embedded dFdCMP using NMR. This structure reveals the following perturbations caused by gemcitabine: (i) the ribose ring of the dFdCMP moiety forms a 3′-endo pucker as opposed to the canonical 2′-endo pucker of a dCMP moiety; (ii) the highly electronegative geminal difluoro group increases the electron density in its vicinity; and (iii) the two fluorine atoms in dFdCMP are physically larger than the corresponding hydrogen atoms in dCMP. These factors are predicted to affect DNA polymerization catalyzed by DNA polymerases, including human pol γ holoenzyme.
Inhibition of DNA Synthesis by Gemcitabine as an Incoming Nucleotide—The pre-steady state kinetic data in Table 2 revealed that relative to dCTP, dFdCTP was incorporated into normal 22/41-mer by pol γ with an 18.5-fold lower kp, whereas the Kd was 23-fold higher, leading to a 432-fold lower kp/Kd value. These kinetic differences can be rationalized as follows. (i) Although dFdCTP is not the same as embedded dFdCMP in DNA, we assume that dFdCTP initially adopts the 3′-endo pucker once it is bound to form the ground-state ternary complex (E·DNA·dNTP). (ii) In order for the phosphodiester bond formation to occur, the conformation of the bound dFdCTP has to be converted to the canonical 2′-endo pucker to allow proper alignment of the primer 3′-OH and the α-phosphate of dFdCTP. (iii) The energy penalty for this conformational conversion should reduce the incorporation rate of dFdCTP. Moreover, the electron-withdrawing difluoro group withdraws electron density from the triphosphate of dFdCTP and thereby reduces the reactivity of the α-phosphate moiety during phosphodiester bond formation. This difluoro group also increases the size of dFdCTP, which may cause a steric clash with the polymerase active site residues. Relative to dCTP, the altered conformation, electrostatics, and physical size likely weakened the interactions between dFdCTP, the template base dGMP, and polymerase active site residues, hence the lower binding affinity of dFdCTP.
Although the efficiency ratio in Table 2 defined dFdCTP as a 432-fold less efficient substrate than dCTP, the incorporation probability of dFdCTP relative to dCTP in vivo, theoretically defined as {[dFdCTP]/[dCTP]}×{(kp/Kd)dFdCTP/(kp/Kd)dCTP}, is likely to be high. Self-potentiation activities of gemcitabine (supplemental Fig. 2) (28, 29) are known to bring about a dramatic cytoplasmic accumulation of dFdCTP, thereby raising the cellular concentration ratio of [dFdCTP]/[dCTP]. For example, Heinemann et al. (26) found that mammalian cells exposed to 100 μm gemcitabine for 4 h showed that cellular pools of dCTP were reduced by 50% and that the cytoplasmic concentration of dFdCTP had risen to over 1 mm. Because relative sizes of individual dNTP pools in the cytosol and mitochondria are similar (45), we expect to find an enriched dFdCTP pool in mitochondria as well. Therefore, it is reasonable to assume that incorporation of dFdCMP into mitochondrial DNA in vivo may be significant.
Compared with FIAUTP, another non-chain-terminating nucleoside analog, dFdCTP is incorporated by pol γ with a 7-fold higher Kd value and a 12-fold slower kp value (Table 2). Comparison of the efficiency ratios for these two nucleotide analogs indicated that dFdCTP is an 86-fold less efficient substrate for human mitochondrial DNA polymerase, which corresponds to and may account for the diminished toxicity of gemcitabine compared with the highly toxic FIAU.
Following dFdCTP incorporation, the DNA primer terminated with 3′-dFdCMP is likely to adopt the 3′-endo pucker conformation as observed in the solution structures of gemcitabine embedded in DNA (44). The electron density of its 3′-OH group should be lowered by the electron-withdrawing fluorine group, thereby rendering the 3′-OH group less nucleophilic. These factors are expected to lower the suitability of the dFdCMP-terminated DNA primer as a substrate for pol γ and hinder the incorporation of the next incoming nucleoside triphosphate. This hypothesis was supported by the observation that correct dGTP was incorporated into 23F/41-mer (Table 1) with a 25-fold smaller kp and a 214-fold lower catalytic efficiency compared with the canonical DNA template 25/45-mer (Table 2) (40). However, the inhibitory effect of a 3′-terminated dFdCMP primer on the catalytic efficiency of extension via correct dNTP incorporation was reduced by 7-fold for the second downstream nucleotide incorporation (Table 2) and was not observed for the third and subsequent nucleotide incorporations (data not shown). Interestingly, human DNA polymerases α and ε, which belong to the B-family, have also been found to inefficiently incorporate dFdCTP. However, in contrast to what has been observed for the A-family enzyme pol γ, these B-family human DNA polymerases exhibit masked chain termination following dFdCMP incorporation. For example, Huang et al. (22) demonstrate that, following the incorporation of a 3′ dFdCMP moiety on a DNA primer, DNA elongation is possible but is very strongly inhibited. In a time period of 1,800 s, only a portion of the primer was extended past the masked termination event, and of those elongation events, only a few dNTPs were added. In contrast, pol γ did not exhibit the expected masked chain termination and was able to extend a DNA primer containing 3′-dFdCMP although with decreased nucleotide incorporation efficiency at the first two downstream positions (Table 2). So far, there are no reported studies on how human DNA polymerases α and ε bypass a template lesion dFdCMP. Interestingly, Fig. 3 showed that within 3 s pol γ was highly successful at bypassing the dFdCMP lesion, albeit in an error-prone manner (Table 4). For wild-type pol γ, only 4% less full-length product was produced in the 3-s time period given a template with embedded dFdCMP (Fig. 3B) compared with the control template (Fig. 3A) (22). Additionally, DNA polymerase θ, the only other member of the A-family DNA polymerases yet identified in humans, has been shown to bypass abasic site and thymine glycol DNA lesions but is completely unable to bypass a variety of DNA lesions resulting from exposure to UV light and cisplatin-based chemotherapy drugs (46). However, the ability of pol θ to bypass dFdCMP or any other nucleoside analog within the context of DNA has yet to be examined.
Incorporated dFdCMP Eludes Editing Mechanism—Upon incorporation at the 3′ DNA primer terminus, the 3′ → 5′-exonuclease activity of pol γ may recognize and excise incorporated dFdCMP as a lesion because of its different conformation and electrostatic character compared with dCMP. Surprisingly, Table 3 revealed that the excision rate of dFdCMP from the DNA primer 3′ terminus (0.0011 s-1) was 55-fold slower than the excision of a corresponding primer terminated with 3′-dCMP (control substrate 23/41-mer). At a nucleotide concentration of 100 μm, pol γ E200A incorporated dGTP into 23F/41-mer with a kp of 1.5 s-1 (Table 2). Based on the principle of kinetic partitioning, the probability of the exonuclease function editing dFdGMP, (kexo/(kexo + kp)) × 100 = (0.0011/(0.0011 + 1.5)) × 100, is calculated to be 0.07%. This probability is much lower than the 80% observed for the correction of a mismatched canonical dNMP (41). Taken together, although the extension of a dFdCMP-terminated primer is 25-fold slower than the extension of a canonical DNA primer, the reduced excision rate constant and thus the extremely low probability of exonuclease editing activity by pol γ increase the likelihood of an incorporated dFdCMP persisting and becoming embedded within the mitochondrial genome if it is not removed by other DNA repair mechanisms. Furthermore, given that mtDNA repair is limited and inefficient (47), persistence of dFdCMP within mtDNA is predicted to be likely. Finally, the toxic side effects of chain-terminating nucleoside analog drugs recede when therapy with the drugs is discontinued (36). However, in the case of FIAU, which has a free 3′-OH group and is not a chain-terminating nucleoside analog, toxicity has been shown to persist and worsen despite immediate withdrawal of the drug, eventually leading to the death of five patients from severe mitochondrial toxicity (48). This suggests that the mitochondria may be able to effectively remove chain-terminating nucleoside analogs and resume normal mtDNA replication, but nucleoside analogs that do not chain terminate, and therefore can become part of the mitochondrial genome, may exert long term toxicity.
Inhibition of Human pol γ-catalyzed DNA Synthesis by Gemcitabine as a Template Base—Fig. 3, B and D, showed that human pol γ holoenzyme, both wild-type and E200A, paused strongly opposite the template base dFdCMP and at the next template position. The kinetic data in Table 4 demonstrated that the efficiency ratio correlated well with the observed pause patterns (Fig. 3). The efficiency ratio (2.2 × 103) reached the poorest value when pol γ attempted to extend the 20/41F-mer, the strongest pause site (Fig. 3, B and D). The second strongest pause site, where pol γ attempted to incorporate dGTP opposite dFdCMP, correlated with the second poorest efficiency ratio value of 1.1 × 103 (Table 4). Our analysis also revealed that, in comparison with the extension of canonical 25/45-mer DNA template, nucleotide incorporation efficiency of pol γ dropped to between 5- and 132-fold at 1 nucleotide preceding dFdCMP and at 2-3 nucleotides downstream of the dFdCMP lesion (Table 4). However, Fig. 3, B and D, did not show obvious pausing by pol γ at these positions. This lack of pausing was because of the relatively high kp values (2-8 s-1) and long reaction times that allowed pol γ to rapidly elongate the corresponding intermediate products. Taken together, one embedded dFdCMP moiety in a DNA template unfavorably affected five nucleotide incorporation events surrounding that lesion. This inhibitory effect may also be due to local DNA structural perturbations caused by dFdCMP (44) as described above. The 3′-endo pucker conformation of template base dFdCMP likely caused unproductive base pairing between an incoming dGTP and dFdCMP, leading to a 188-fold higher Kd value for the binding of dGTP to pol γ·19/41F-mer (Table 4). Similarly, dFd-CMP also decreased the binding affinity of the next 2 downstream nucleotides by 6-10-fold before recovering to normal. The predicted decrease in the structural integrity of the DNA duplex caused by the presence of a dFdCMP moiety with a 3′-endo pucker is supported by the fact that the melting temperature of the 12-bp Okazaki fragment is lowered by 4.3 °C in the presence of an internal dFdCMP (44). Such a noncanonical conformation of dFdCMP in a template should also affect the positioning of an incoming dNTP for in-line attack by the primer's 3′-OH group during phosphodiester bond formation. If this indeed is occurring, it would readily explain the reduction in kp values observed. The negative impact was largest (227-fold) for dTTP incorporation into 20/41F-mer. Interestingly, the inhibitory effect of gemcitabine was larger as a template base than as an incoming nucleotide when we compared the efficiency ratios in Tables 2 and 4. This suggested that if a ribose pucker conversion is necessary for correct orientation of the nucleotide triphosphate prior to the chemistry step, it may be more difficult when gemcitabine is constrained within the template sequence than when it is either an incoming nucleotide triphosphate or a 3′-primer terminal base. However, the reason for these interesting observations cannot be unambiguously determined here.
Unfaithful Bypass of Template dFdCMP—When a template lesion causes a DNA polymerase to pause, the enzyme tends to catalyze 5′ → 3′ DNA polymerization in an error-prone manner. This general trend was found to hold true when pol γ bypassed the template dFdCMP moiety. Table 5 shows that the polymerase activity of pol γ has a fidelity of 3.7 × 102 to 3.0 × 103 at the two strong pause sites (Fig. 3, B and D), which is significantly lower than the corresponding fidelity of 2.8 × 105 to 6.4 × 105 determined with canonical DNA substrate 25/45-mer (43). Thus, pol γ was 200-800-fold less faithful upon encountering a dFdCMP template lesion, thus promoting DNA mutagenesis. To compound these mutagenic events, the misincorporated canonical nucleotide (for example 3′-dTMP in 20T/41F-mer) was excised more slowly than a single 3′-mismatched primer base in a canonical DNA template (40). In addition, the matched primer terminal 3′-dGMP in 20/41F-mer was removed at half the speed of another correctly paired, canonical 3′-primer terminal dNMP (23/41-mer substrate, Table 3). Therefore, the inhibited exonuclease activity as a result of the template-embedded dFdCMP may actually facilitate nucleotide misincorporation by slowing the 3′ → 5′-exonuclease activity of pol γ. With respect to the editing function of pol γ, the currently established mechanism is that the primer 3′-mis-matched base is transferred from the polymerase active site to the 3′ → 5′-exonuclease active site for excision, whereas the template strand remains at the polymerase active site of pol γ (41). With this in mind, there arise at least two possible explanations for the observed exonucleolytic rate. Either the presence of dFdCMP in the template directly inhibited the exonucleolytic activity or it hindered the transfer of the 3′-primer terminus to the proofreading active site or possibly both. More studies in our laboratory are under way to examine these possibilities.
Potential Relevance to Observed Gemcitabine Clinical Toxicity—Our kinetic analysis directly confirmed that gemcitabine, as both an incoming nucleotide and as a template base, inhibited DNA synthesis catalyzed by human pol γ. Moreover, our studies also revealed that each template dFdCMP was a mutagenic “hot spot” during DNA replication. Additionally, mutagenic effects may be exerted by gemcitabine metabolites within the mitochondria. For instance, it is known that gemcitabine metabolites cause cellular dNTP pool imbalances by inhibiting ribonucleotide reductase (23, 24), CTP synthetase (25, 26), and dCMP deaminase (27). Therefore, gemcitabine therapy is also likely to cause an imbalance of mitochondrial nucleotide pools. Such imbalances have been found previously to be mutagenic to the mitochondrial genome (49) and lead to disease states such as mitochondrial neurogastrointestinal encephalomyopathy (50). Considering that mammalian cells have 1,000-5,000 copies of the mitochondrial genome (51, 52) and given that mtDNA replication occurs continuously throughout the entire cell cycle (45, 53), these inhibitory and mutagenic effects of gemcitabine may result in growing mitochondrial genomic instability over time and, like FIAU, may be persistent.
Supplementary Material
Acknowledgments
We acknowledge the contributions of both Harold Lee and Chris Hamilton for preparing human DNA polymerase γ.
This work was supported, in whole or in part, by National Institutes of Health Grants GM079403 (to Z. S.) and GM044613 (to K. A. J.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1-3.
Footnotes
The abbreviations used are: dFdC, 2′-deoxy-2′,2′-difluorocytidine; dFdCMP, gemcitabine 5′-monophosphate; dFdCDP, gemcitabine 5′-diphosphate; dFdCTP, gemcitabine 5′-triphosphate; FIAU, 1-(2-deoxy-2-fluoro-β-d-arabinofuranosyl)-5-iodouracil; pol γ, human DNA polymerase γ holoenzyme; mtDNA, mitochondrial DNA; SSU, human mtDNA polymerase small subunit.
References
- 1.Crino, L., Scagliotti, G. V., Ricci, S., De Marinis, F., Rinaldi, M., Gridelli, C., Ceribelli, A., Bianco, R., Marangolo, M., Di Costanzo, F., Sassi, M., Barni, S., Ravaioli, A., Adamo, V., Portalone, L., Cruciani, G., Masotti, A., Ferrara, G., Gozzelino, F., and Tonato, M. (1999) J. Clin. Oncol. 17 3522-3530 [DOI] [PubMed] [Google Scholar]
- 2.Burris, H. A., III, Moore, M. J., Andersen, J., Green, M. R., Rothenberg, M. L., Modiano, M. R., Cripps, M. C., Portenoy, R. K., Storniolo, A. M., Tarassoff, P., Nelson, R., Dorr, F. A., Stephens, C. D., and Von Hoff, D. D. (1997) J. Clin. Oncol. 15 2403-2413 [DOI] [PubMed] [Google Scholar]
- 3.Morandi, P. (2006) Ann. Oncol. 17 Suppl. 5, V177-V180 [DOI] [PubMed] [Google Scholar]
- 4.Lorusso, D., Di Stefano, A., Fanfani, F., and Scambia, G. (2006) Ann. Oncol. 17 Suppl. 5, V188-V194 [DOI] [PubMed] [Google Scholar]
- 5.Sallah, S., Wan, J. Y., and Nguyen, N. P. (2001) Br. J. Haematol. 113 185-187 [DOI] [PubMed] [Google Scholar]
- 6.Einhorn, L. H., Stender, M. J., and Williams, S. D. (1999) J. Clin. Oncol. 17 509-511 [DOI] [PubMed] [Google Scholar]
- 7.Kubicka, S., Rudolph, K. L., Tietze, M. K., Lorenz, M., and Manns, M. (2001) Hepatogastroenterology 48 783-789 [PubMed] [Google Scholar]
- 8.Catimel, G., Vermorken, J. B., Clavel, M., de Mulder, P., Judson, I., Sessa, C., Piccart, M., Bruntsch, U., Verweij, J., Wanders, J., Franklin, H., Kaye, S. B., and the EORTC Early Clinical Trials Group (1994) Ann. Oncol. 5 543-547 [DOI] [PubMed] [Google Scholar]
- 9.Mutch, D. G., and Bloss, J. D. (2003) Gynecol. Oncol. 90 S8-S15 [DOI] [PubMed] [Google Scholar]
- 10.Dumontet, C., Morschhauser, F., Solal-Celigny, P., Bouafia, F., Bourgeois, E., Thieblemont, C., Leleu, X., Hequet, O., Salles, G., and Coiffier, B. (2001) Br. J. Haematol. 113 772-778 [DOI] [PubMed] [Google Scholar]
- 11.Dalbagni, G., Russo, P., Bochner, B., Ben-Porat, L., Sheinfeld, J., Sogani, P., Donat, M. S., Herr, H. W., and Bajorin, D. (2006) J. Clin. Oncol. 24 2729-2734 [DOI] [PubMed] [Google Scholar]
- 12.Fracasso, P. M., Tan, B. R., Jr., Grieff, M., Stephenson, J., Jr., Liapis, H., Umbeck, N. L., Von Hoff, D. D., and Rowinsky, E. K. (1999) J. Natl. Cancer Inst. 91 1779-1780 [DOI] [PubMed] [Google Scholar]
- 13.Pfisterer, J., Plante, M., Vergote, I., du Bois, A., Hirte, H., Lacave, A. J., Wagner, U., Stahle, A., Stuart, G., Kimmig, R., Olbricht, S., Le, T., Emerich, J., Kuhn, W., Bentley, J., Jackisch, C., Luck, H. J., Rochon, J., Zimmermann, A. H., and Eisenhauer, E. (2006) J. Clin. Oncol. 24 4699-4707 [DOI] [PubMed] [Google Scholar]
- 14.Airoldi, M., Cattel, L., Passera, R., Pedani, F., Milla, P., and Zanon, C. (2006) Pancreas 32 44-50 [DOI] [PubMed] [Google Scholar]
- 15.Shirai, T., Hirose, T., Noda, M., Ando, K., Ishida, H., Hosaka, T., Ozawa, T., Okuda, K., Ohnishi, T., Ohmori, T., Horichi, N., and Adachi, M. (2006) Lung Cancer 52 181-187 [DOI] [PubMed] [Google Scholar]
- 16.Mackey, J. R., Mani, R. S., Selner, M., Mowles, D., Young, J. D., Belt, J. A., Crawford, C. R., and Cass, C. E. (1998) Cancer Res. 58 4349-4357 [PubMed] [Google Scholar]
- 17.Mackey, J. R., Yao, S. Y., Smith, K. M., Karpinski, E., Baldwin, S. A., Cass, C. E., and Young, J. D. (1999) J. Natl. Cancer Inst. 91 1876-1881 [DOI] [PubMed] [Google Scholar]
- 18.Marce, S., Molina-Arcas, M., Villamor, N., Casado, F. J., Campo, E., Pastor-Anglada, M., and Colomer, D. (2006) Haematologica 91 895-902 [PubMed] [Google Scholar]
- 19.Blackstock, A. W., Lightfoot, H., Case, L. D., Tepper, J. E., Mukherji, S. K., Mitchell, B. S., Swarts, S. G., and Hess, S. M. (2001) Clin. Cancer Res. 7 3263-3268 [PubMed] [Google Scholar]
- 20.Richardson, K. A., Vega, T. P., Richardson, F. C., Moore, C. L., Rohloff, J. C., Tomkinson, B., Bendele, R. A., and Kuchta, R. D. (2004) Biochem. Pharmacol. 68 2337-2346 [DOI] [PubMed] [Google Scholar]
- 21.Ruiz van Haperen, V. W., Veerman, G., Vermorken, J. B., and Peters, G. J. (1993) Biochem. Pharmacol. 46 762-766 [DOI] [PubMed] [Google Scholar]
- 22.Huang, P., Chubb, S., Hertel, L. W., Grindey, G. B., and Plunkett, W. (1991) Cancer Res. 51 6110-6117 [PubMed] [Google Scholar]
- 23.Shao, J., Zhou, B., Chu, B., and Yen, Y. (2006) Curr. Cancer Drug Targets 6 409-431 [DOI] [PubMed] [Google Scholar]
- 24.Plunkett, W., Huang, P., Searcy, C. E., and Gandhi, V. (1996) Semin. Oncol. 23 3-15 [PubMed] [Google Scholar]
- 25.Davidson, J. D., Ma, L., Flagella, M., Geeganage, S., Gelbert, L. M., and Slapak, C. A. (2004) Cancer Res. 64 3761-3766 [DOI] [PubMed] [Google Scholar]
- 26.Heinemann, V., Schulz, L., Issels, R. D., and Plunkett, W. (1995) Semin. Oncol. 22 11-18 [PubMed] [Google Scholar]
- 27.Neff, T., and Blau, C. A. (1996) Exp. Hematol. 24 1340-1346 [PubMed] [Google Scholar]
- 28.Plunkett, W., Huang, P., Xu, Y. Z., Heinemann, V., Grunewald, R., and Gandhi, V. (1995) Semin. Oncol. 22 3-10 [PubMed] [Google Scholar]
- 29.Heinemann, V., Xu, Y. Z., Chubb, S., Sen, A., Hertel, L. W., Grindey, G. B., and Plunkett, W. (1992) Cancer Res. 52 533-539 [PubMed] [Google Scholar]
- 30.Verstappen, C. C., Postma, T. J., Hoekman, K., and Heimans, J. J. (2003) J. Neurooncol. 63 201-205 [DOI] [PubMed] [Google Scholar]
- 31.Dormann, A. J., Grunewald, T., Wigginghaus, B., and Huchzermeyer, H. (1998) Lancet 351 644. [DOI] [PubMed] [Google Scholar]
- 32.Lewis, W., and Dalakas, M. C. (1995) Nat. Med. 1 417-422 [DOI] [PubMed] [Google Scholar]
- 33.Semino-Mora, C., Leon-Monzon, M., and Dalakas, M. C. (1997) Lab. Investig. 76 487-495 [PubMed] [Google Scholar]
- 34.Brahams, D. (1994) Lancet 343 1494-1495 [DOI] [PubMed] [Google Scholar]
- 35.Johnson, A. A., Tsai, Y., Graves, S. W., and Johnson, K. A. (2000) Biochemistry 39 1702-1708 [DOI] [PubMed] [Google Scholar]
- 36.Johnson, A. A., Ray, A. S., Hanes, J., Suo, Z., Colacino, J. M., Anderson, K. S., and Johnson, K. A. (2001) J. Biol. Chem. 276 40847-40857 [DOI] [PubMed] [Google Scholar]
- 37.Graves, S. W., Johnson, A. A., and Johnson, K. A. (1998) Biochemistry 37 6050-6058 [DOI] [PubMed] [Google Scholar]
- 38.Roettger, M. P., Fiala, K. A., Sompalli, S., Dong, Y., and Suo, Z. (2004) Biochemistry 43 13827-13838 [DOI] [PubMed] [Google Scholar]
- 39.Fiala, K. A., Duym, W. W., Zhang, J., and Suo, Z. (2006) J. Biol. Chem. 281 19038-19044 [DOI] [PubMed] [Google Scholar]
- 40.Johnson, A. A., and Johnson, K. A. (2001) J. Biol. Chem. 276 38090-38096 [DOI] [PubMed] [Google Scholar]
- 41.Johnson, A. A., and Johnson, K. A. (2001) J. Biol. Chem. 276 38097-38107 [DOI] [PubMed] [Google Scholar]
- 42.Johnson, K. A. (1992) Enzymes 20 1-61 [Google Scholar]
- 43.Lee, H. R., and Johnson, K. A. (2006) J. Biol. Chem. 281 36236-36240 [DOI] [PubMed] [Google Scholar]
- 44.Konerding, D., James, T. L., Trump, E., Soto, A. M., Marky, L. A., and Gmeiner, W. H. (2002) Biochemistry 41 839-846 [DOI] [PubMed] [Google Scholar]
- 45.Ferraro, P., Nicolosi, L., Bernardi, P., Reichard, P., and Bianchi, V. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 18586-18591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ukai, A., Maruyama, T., Mochizuki, S., Ouchida, R., Masuda, K., Kawamura, K., Tagawa, M., Kinoshita, K., Sakamoto, A., Tokuhisa, T., and O-Wang, J. (2006) Genes Cells 11 111-121 [DOI] [PubMed] [Google Scholar]
- 47.Bogenhagen, D. F. (1999) Am. J. Hum. Genet. 64 1276-1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McKenzie, R., Fried, M. W., Sallie, R., Conjeevaram, H., Di Bisceglie, A. M., Park, Y., Savarese, B., Kleiner, D., Tsokos, M., Luciano, C., Pruett, T., Stotka, J. L., Straus, S. E., and Hoofnagle, J. H. (1995) N. Engl. J. Med. 333 1099-1105 [DOI] [PubMed] [Google Scholar]
- 49.Song, S., Wheeler, L. J., and Mathews, C. K. (2003) J. Biol. Chem. 278 43893-43896 [DOI] [PubMed] [Google Scholar]
- 50.Nishino, I., Spinazzola, A., and Hirano, M. (1999) Science 283 689-692 [DOI] [PubMed] [Google Scholar]
- 51.Bogenhagen, D., and Clayton, D. A. (1974) J. Biol. Chem. 249 7991-7995 [PubMed] [Google Scholar]
- 52.Shmookler Reis, R. J., and Goldstein, S. (1983) J. Biol. Chem. 258 9078-9085 [PubMed] [Google Scholar]
- 53.Bogenhagen, D., and Clayton, D. A. (1977) Cell 11 719-727 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.