

Abstract
We previously showed that agmatine stimulated hepatic ureagenesis. In this study, we sought to determine whether the action of agmatine is mediated via cAMP signaling. A pilot experiment demonstrated that the phosphodiesterase inhibitor, 3-isobutylmethylxanthine (IBMX), inhibited urea synthesis albeit increased [cAMP]. Thus, we hypothesized that IBMX inhibits hepatic urea synthesis independent of [cAMP]. We further theorized that agmatine would negate the IBMX action and improve ureagenesis. Experiments were carried out with isolated mitochondria and 15NH4Cl to trace [15N]citrulline production or [5-15N]glutamine and a rat liver perfusion system to trace ureagenesis. The results demonstrate that IBMX induced the following: (i) inhibition of the mitochondrial respiratory chain and diminished O2 consumption during liver perfusion; (ii) depletion of the phosphorylation potential and overall hepatic energetic capacity; (iii) inhibition of [15N]citrulline synthesis; and (iv) inhibition of urea output in liver perfusion with little effect on [N-acetylglutamate]. The results indicate that IBMX directly and specifically inhibited complex I of the respiratory chain and carbamoyl-phosphate synthase-I (CPS-I), with an EC50 about 0.6 mm despite a significant elevation of hepatic [cAMP]. Perfusion of agmatine with IBMX stimulated O2 consumption, restored hepatic phosphorylation potential, and significantly stimulated ureagenesis. The action of agmatine may signify a cascade effect initiated by increased oxidative phosphorylation and greater ATP synthesis. In addition, agmatine may prevent IBMX from binding to one or more active site(s) of CPS-I and thus protect against inhibition of CPS-I. Together, the data may suggest a new experimental application of IBMX in studies of CPS-I malfunction and the use of agmatine as intervention therapy.
It is well established that hepatic ureagenesis is up-regulated by cAMP, the second messenger of glucagon (1, 2). We previously reported that agmatine (AGM),2 the product of arginine decarboxylase (3), mimics the action of glucagon on hepatic metabolism, including stimulation of fatty acid oxidation (FAO), increasing oxygen consumption, and augmentation of N-acetylglutamate (NAG), and thus urea synthesis in a liver perfusion system (4, 5). Our data suggest that the agmatine action may be mediated via elevation of cellular [cAMP] (5). Indeed, we subsequently demonstrated that AGM when given orally in drinking water elevated [cAMP] in rat kidney cortex (6).
In most cells and tissues the level of cAMP depends on the balance between the respective rates of synthesis and hydrolysis. The maximal rate of cAMP synthesis is less than one-tenth the maximal rate of hydrolysis by phosphodiesterases (PDEs) (7), suggesting that the cAMP concentration and activation of cAMP-dependent protein kinase signaling pathways depend in large measure upon the capacity of PDEs. Many studies have utilized 3-isobutylmethylxanthine (IBMX) to elevate cellular cAMP. The commonly employed concentration of this nonspecific inhibitor of PDEs was between 0.5 and 1 mm (7).
In this study, we sought to further characterize the effect of AGM on hepatic [cAMP] and thereby ureagenesis with or without concomitant supplementation of IBMX. Pilot experiments indicated that IBMX significantly inhibited ureagenesis during liver perfusion. The down-regulation of ureagenesis despite increased cellular cAMP concentration may represent “an anomaly” in the regulation of hepatic ureagenesis. Therefore, the goal of this work is to explore this unanticipated effect of IBMX, which is widely used as a PDE inhibitor in studies of second messenger systems and signal transduction pathways. A detailed study of IBMX action may shed new light on the regulation of hepatic ureagenesis by cAMP.
A major hepatic function is conversion of ammonia to urea (1). The initial step in urea synthesis is the conversion of NH+4 and HCO–3 into carbamoyl phosphate (CP) via mitochondrial carbamoyl-phosphate synthase-I (CPS-I) (EC 6.3.4.16) (1, 8–12), which requires NAG as an obligatory activator (13). CP is combined with ornithine via the ornithine transcarbamylase (OTC) reaction to form citrulline (1). It is believed that the main regulatory site of urea synthesis is mitochondrial CPS-I, which requires two ATPs for the synthesis of CP (1). Thus, we hypothesized that IBMX may inhibit CPS-I by lowering hepatic oxidative phosphorylation (OXPHOS) and energetic capacity. We further hypothesized that AGM would negate the IBMX-induced inhibition of urea synthesis. Our rationale is based on our prior findings that AGM protects against drug-induced inhibition of complex I and oxidative phosphorylation (6). In addition, AGM was found to stimulate the following: (i) FAO; (ii) the incorporation of acetyl-CoA into the tricarboxylic acid cycle; and (iii) oxygen consumption (5). The stimulation of FAO provided more acetyl-CoA for NAG synthesis (5) as well as increased pyruvate carboxylase and thus [oxaloacetate] (14, 15). The latter is used for the synthesis of aspartate, which contributes the second nitrogen for urea synthesis (16, 17).
We therefore designed experiments to determine the following: (i) the site(s) of IBMX-induced inhibition of urea synthesis; (ii) whether the IBMX action on ureagenesis is coupled with other pathways such as oxidative phosphorylation and/or phosphorylation potential and energetic capacity; and (iii) whether supplementation of AGM with IBMX during liver perfusion will ameliorate the IBMX-induced inhibition of ureagenesis. To address these questions, experiments were carried out with either a liver perfusion system or isolated mitochondria. We used 15N-labeled ammonia or [5-15N]glutamine to assess the production of NAG and CP and thus the rates of urea formation (5, 18, 19). Because the process of urea synthesis involves equimolar consumption of NH+4 and aspartate-N, and because glutamine is a major source of ammonia and aspartate-N for urea synthesis (16–18), the use of [5-15N]glutamine was meant to determine in liver perfusion the rate of ureagenesis in a physiological setting, as demonstrated previously (4, 17, 18). Isolated mitochondria and/or mitochondrial extracts were used to determine the synthesis of [15N]citrulline from 15NH4Cl and bicarbonate. The production of [15N]citrulline was used as a proxy for the production of CP, and thus, activity of CPS-I as indicated previously (8–12). Isolated mitochondria were also used to examine the action of IBMX on mitochondrial respiratory chain complexes and thus OXPHOS.
A separate series of experiments was carried out to determine whether the action of AGM on OXPHOS is coupled with pyruvate dehydrogenase (PDH) or the tricarboxylic acid cycle activity. Pyruvate is metabolized via the PDH complex to form NADH and acetyl-CoA (5, 20). The incorporation and metabolism of acetyl-CoA in the tricarboxylic acid cycle produce more NADH as well as FADH2 for mitochondrial OXPHOS.
EXPERIMENTAL PROCEDURES
Materials and Animals—Male Sprague-Dawley rats (Charles River Breeding Laboratories) were fed ad libitum a standard rat chow diet. Chemicals were of analytical grade and obtained from Sigma. Enzymes and cofactors for the analysis of adenine nucleotides, urea, lactate, pyruvate, and ammonia were obtained from Sigma. U-13C3-Labeled Pyruvate and -lactate, 15NH4Cl, or [5-15N]glutamine, 99 mol % excess, were from Isotec.
Experiments with Liver Perfusion—Livers from overnight fasted male rats were perfused in the non-recirculating mode and antegrade flow at a rate of 3–3.5 ml/g liver/min, as described previously (4, 5). The basic perfusion medium was Krebs saline, pH 7.4, continuously gassed with 95% O2, 5% CO2 and containing lactate (2.1 mm) and pyruvate (0.3 mm) as metabolic fuels. pO2 (in influent and effluent media) was monitored throughout, and oxygen consumption was calculated. After 15 min of conditioning with a basic perfusion medium, the perfusate was replaced with one containing U-13C3-labeled lactate (2.1 mm), -pyruvate (0.3 mm), NH4Cl (0.3 mm), and [5-15N]glutamine (1 mm), with or without 0.5 mm IBMX, dissolved in DMSO or IBMX plus 0.1 mm agmatine. Control perfusion was carried out with DMSO. The perfusion was continued for an additional 45 min. Samples were taken from the influent and effluent media for chemical and isotopic analyses. At the end of perfusion, the liver was freeze-clamped with aluminum tongs pre-cooled in liquid N2. The frozen liver was ground into a fine powder, extracted into perchloric acid, neutralized, and used for determination of metabolite level and 13C or 15N enrichment.
Studies with Isolated Mitochondria—Isolated intact mitochondria or freeze-thawed broken mitochondria were used to explore the action of IBMX on the mitochondrial function. Mitochondria were isolated from the liver of overnight fasted rats by differential centrifugation as described previously (5, 19). Respiratory control and oxygen consumption were determined (5, 6, 19). Metabolic studies were carried out with mitochondria having a V3/V2 ratio greater than 3.
In the first series of experiments, we determined the action of IBMX on the synthesis of 15N-citrulline from 15N-labeled ammonia. Synthesis of citrulline has been used as a proxy for CP production (8–12). Mitochondrial suspensions (≈2 mg of protein·ml–1) were incubated in Erlenmeyer flasks (2 ml final volume) at 30 °C in a shaking water bath for 20 min in a basic incubation medium consisting of the following (in mm): 50 Tris, 2 EDTA, 5 KCl, 5 MgCl2, 15 KHCO3, and 5 KH2PO4, pH 7.4. Incubations were carried out with increasing concentrations of IBMX (dissolved in DMSO), ATP, and various 15N-labeled and unlabeled substrates of carbamoyl phosphate synthesis.
In separate experiments, a freeze-thawed and sonicated mitochondrial extract served as a source of either OTC or carbamoyl-phosphate synthase-I, as indicated (8). The action of IBMX on the activity of OTC was determined by incubation of a mitochondrial extract with basic medium containing the following (in mm): 10 carbamoyl phosphate, 10 ornithine, 5 ATP, and increasing concentrations (0–5 mm) of IBMX. Parallel experiments were carried out with commercially available (Sigma) OTC. For determination of CPS-I activity, the mitochondrial extract was incubated with the following (in mm): 10 15NH4Cl, 10 ornithine, 5 ATP, 1 N-acetylglutamate, and increasing concentrations (0–5 mm) of IBMX.
To examine the effect of IBMX on oxidative phosphorylation, mitochondrial suspensions in basic medium were supplemented with the following: either 5 mm malate plus 5 mm glutamate, which formed NADH that is oxidized in complex I; 5 mm succinate, which formed FADH2 that is oxidized in complex II; or 0.24 mm N,N,N,N-tetramethyl-p-phenylenediamine (TMPD) plus 7.2 mm ascorbate that is oxidized in complex IV (6, 21). After recording of state 2, 0.3 mm ADP was added, and state 3 and state 4 were determined. In a separate series of measurements, carbonyl cyanide m-chlorophenylhydrazone (ClCCP) was used to uncouple respiration from phosphorylation. Following determination of ADP-dependent respiration without the addition of ClCCP, the uncoupled respiration was determined after addition of 10 μm ClCCP.
A separate series of experiment was carried out to determine the flux through the PDH or the tricarboxylic acid cycle activity. To examine the action of IBMX on the PDH reaction, mitochondria were incubated with basic medium plus 5 mm [1-13C]pyruvate. To monitor the incorporation of acetyl-CoA into the tricarboxylic acid cycle, mitochondria were incubated with basic medium plus 5 mm [U-13C]pyruvate. Isolated mitochondria were incubated for 20 min. 13CO2 was collected and analyzed for 13C enrichment as indicated (5, 6).
At the end of the incubation, an aliquot (100 μl) was taken for protein determination, and the incubation was stopped with 100–150 μl of HClO4 (60%). Metabolite measurements were done in neutralized extracts. Three to five independent experiments were carried out per each series.
GC-MS and NMR Methodology—GC-MS measurements of 13C or 15N isotopic enrichment were performed on either a Hewlett Packard 5970 mass selective detector (MSD) and/or 5971 MSD, coupled with a 5890 HP-GC, GC-MS Agilent system (6890 GC-5973 MSD) or Hewlett-Packard HP-5970 MSD, using electron impact ionization with an ionizing voltage of –70 eV and an electron multiplier set to 2000 V.
For measurement of 13Cor 15N enrichment in amino acids or urea, the liver extract or effluent samples were prepared as described previously (5, 19). 15N enrichment in effluent ammonia was determined as in Ref. 17. In addition, a portion (about 1 g wet weight) of the neutralized liver extract was analyzed by 13C NMR or 31P NMR methodology using a Bruker Avance DMX 400 wide bore equipped with a Hewlett-Packard computer (22). The chemical shift of analyte containing 13C was determined relative to the resonance of trimethylsilylpropionic acid at –2.7 ppm. The chemical shifts of inorganic phosphate (Pi) and adenine nucleotides relative to the resonance of methylene diphosphonate at 16.7 ppm were measured as described (23). Data acquisition and calculation of % 13C enrichment in various carbons were determined as described (22).
The concentration and isotopic enrichment in NAG in liver or mitochondrial extracts were determined using GC-MS and isotope dilution (5, 19). The formation of NAG isotopomers was monitored using ions at m/z 158, 159, 160, 161, 162 and 163, for M + 1, M +2, M + 3, M + 4, and M + 5 (containing 1–5 13C atoms), respectively. In experiments with isolated mitochondria, the production of 15N-labeled NAG and citrulline from 15N-labeled precursors was determined as described (5, 19).
The production of 13CO2 following mitochondrial incubation with [1-13C]pyruvate or [U-13C]pyruvate was monitored as in Ref. 6. Briefly, the CO2 released was trapped in center wells with 250 μl of 0.2 mm NaOH. At the end of incubation 10 μl of NaOH was removed and transferred into a sealed tube containing 1 ml of 1 mm NaHCO3. Then 100 μl of 20% phosphoric acid was added and left for about 30 min to liberate CO2. The latter was removed with a sealed syringe and transferred to auto-sampler tubes for analysis. Isotopic enrichment in 13CO2 was determined by an isotope ratio-mass spectrometer (Thermoquest Finnigan Delta Plus), using the m/z 45/44 ratio as indicated (5, 6).
Other Assays—The concentration of amino acids was determined by HPLC, utilizing pre-column derivatization with o-phthalaldehyde (24). The levels of ammonia and urea were measured (4, 19). ATP (25), ADP, and AMP were determined as in Ref. 26. Adenine nucleotides were also determined by 31P NMR as indicated above. cAMP was measured as described (27) using a cAMP EIA kit (Cayman Chemical Co.).
Calculations and Statistical Analyses—During liver perfusion, the rate of uptake or the output of metabolites was determined by the measurement of metabolite concentration in the influent and effluent (nmol/ml), normalized to the flow rate (ml/min) and liver wet weight (5, 19). 15N enrichment (mol % excess) in amino acids or urea was calculated as indicated (18, 19). The output of 15N-labeled urea isotopomers, that is Um+1 and Um+2, urea labeled in one or two N atoms, respectively, was determined as described (28) and used for calculation of the flux through the phosphate-dependent glutaminase (PDG) (18). In experiments with isolated mitochondria, the release of 13CO2 (nmol/mg protein) from [1-13C]pyruvate should represent the flux through PDH. The subtraction of 13CO2 release in experiments with [U-13C]pyruvate from that in experiments with[1-13C]pyruvate represents the oxidation of acetyl-CoA in the tricarboxylic acid cycle (6). Data obtained from mitochondrial incubations were analyzed with GraphPad Prism-4 software for linear and nonlinear curve fitting.
Each series of experiments was repeated 3–4 times with different mitochondrial preparations or with individual liver perfusion systems as outlined above. Statistical analysis was carried out using InSTAT 1.14 software for the Macintosh. The Student's t test or analysis of variance test was employed to compare two groups or differences among groups as needed. A p value less than 0.05 was taken as indicating a statistically significant difference.
RESULTS
Metabolic Profile of the Liver Following IBMX or IBMX + AGM Perfusions—Data in Table 1 represent the metabolic profile of freeze-clamped livers in the control state or following infusion with IBMX or IBMX + AGM. There was a significant increase in [cAMP] in livers perfused with IBMX or IBMX + AGM compared with control (Table 1). Perfusion with only IBMX decreased O2 consumption by 20–30%. The addition of AGM with IBMX significantly increased O2 consumption compared with perfusion with only IBMX (Table 1). The content of adenine nucleotides was similar to levels found previously (5, 18, 19). [ATP] was decreased by about 30% following perfusion with IBMX, and it increased by about 30% when AGM was added. The changes in [ATP] are not significant compared with control perfusion, but [ATP] was significantly higher with IBMX plus AGM compared with perfusion with only IBMX (Table 1). Calculation of phosphorylation potential ([ATP]/[ADP] × [Pi]) (29), indicates a significant decrease with IBMX. The supplementation of AGM with IBMX reversed the decrease in phosphorylation potential. The latter was significantly higher in IBMX + AGM compared with IBMX perfusions but not changed compared with control (Table 1). The data indicate that the changes in O2 consumption were tightly linked with [ATP] as well as the phosphorylation potential (Table 1).
TABLE 1.
Metabolic state of the liver at the end of perfusions in control, with IBMX, or with IBMX + agmatine
Livers from overnight fasted male rats were perfused in the non-recirculating mode without IBMX (Control), with IBMX (0.5 mm), or with IBMX (0.5 mm) + agmatine (0.1 mm) and lactate, pyruvate, glutamine, and ammonia (as described under “Experimental Procedures”). At the end of the perfusion, the liver was freeze-clamped, extracted into perchloric acid, and used for metabolite determination.
| Control | IBMX | IBMX + agmatine | |
|---|---|---|---|
| I. Oxygen consumptiona (μmol·g-1·min-1) | 1.9 ± 0.2 | 1.6 ± 0.2 | 2.2 ± 0.1b |
| II. cAMP (nmol·g-1) | 3.15 ± 0.9 | 8.4 ± 2.9c | 5.2 ± 0.8c |
| III. Adenine nucleotides and Pi (μmol·g-1) and phosphorylation potential | |||
| ATP | 3.4 ± 0.9 | 2.7 ± 0.7 | 4.5 ± 1.1b |
| ADP | 1.6 ± 0.4 | 1.8 ± 0.6 | 2.1 ± 0.1 |
| AMP | 0.49 ± 0.06 | 0.39 ± 0.1 | 0.51 ± 0.08 |
| Pi | 5.6 ± 0.9 | 5.6 ± 1.1 | 6.1 ± 0.4 |
| Phosphorylation potentiald | 0.37 ± 0.06 | 0.28 ± 0.03c | 0.41 ± 0.08b |
| IV. Urea cycle (nmol·g-1) | |||
| Citrulline | 120 ± 38 | 74 ± 19c | 103 ± 29 |
| N-Acetylglutamate | 38 ± 8 | 39 ± 5 | 41 ± 3 |
The rates of oxygen consumption are the steady-state values obtained between 30 and 60 min after the start of infusion. Values are mean ± S.D. of 4-5 perfusions.
Indicates significant difference (p < 0.05) compared with perfusion with IBMX.
Indicates significant difference (p < 0.05) compared with control.
Calculated as described in Ref. 29. Levels of adenine nucleotides, Pi, citrulline, and N-acetylglutamate are means ± S.D. of separate livers (n = 3-4).
Analysis of intermediary metabolites in freeze-clamped liver at the end of perfusions with IBMX or IBMX + AGM showed insignificant differences in the concentration of citrate, malate, succinate, or α-ketoglutarate compared with control. Similarly, IBMX had little effect on the level of alanine, aspartate, glutamate, or glutamine, the main substrates for urea-N (data not shown). An observation of special importance is that IBMX had no effect on the level of N-acetylglutamate, the activator of CPS-I (13); however, IBMX significantly decreased the level of citrulline (Table 1), the chief mitochondrial intermediate of the hepatic urea cycle (1). The addition of AGM attenuated this decrease in citrulline concentration (Table 1). Because the citrulline level is a marker for carbamoyl phosphate generation via CPS-I (8–12), the striking fall in citrulline concentration may reflect inhibition of carbamoyl phosphate synthesis, and thereby urea production.
The Action of IBMX or IBMX + AGM on Hepatic Ureagenesis during Liver Perfusion—To explore the hypothesis that IBMX inhibits ureagenesis despite the elevation of [cAMP], liver perfusions were performed with [5-15N]glutamine and unlabeled ammonia as detailed under “Experimental Procedures.” Data in Fig. 1A demonstrate that the output of total urea-N from perfusate glutamine and ammonia was decreased by about 50% (p < 0.05) in perfusions with IBMX. The addition of AGM with IBMX significantly increased urea output. Notwithstanding the remarkable decrease in total urea output, the data in Fig. 1, B and C, demonstrate insignificant changes in 15N enrichment (mol % excess) in the Um+1 and Um+2 isotopomers in perfusions with IBMX or IBMX + AGM compared with control, indicating that neither IBMX nor supplementation of AGM affected the fractional contribution of amide-N of glutamine to urea synthesis.
FIGURE 1.

The effect of IBMX or IBMX + AGM on the output of total and [15N]urea during the course of perfusion. Pre-perfusions (15 min) were carried out with basic perfusate containing the following (in mm): 2.1 lactate and 0.3 pyruvate. Liver then was perfused for an additional 45 min with perfusate containing the following (in mm): 0.5 IBMX, 0.5 IBMX + AGM 0.1, 1 [5-15N]glutamine, 0.3 NH4Cl, 2.1 lactate, and 0.3 pyruvate. A, represents total urea-N output; B, represents the 15N enrichment in urea containing one 15N (M +1); and C represents the 15N enrichment in urea containing two 15N atoms (M + 2). Bars are mean ± S.D. of 3–4 independent liver perfusions.
The amide-N of glutamine is the major source of urea-N (4, 18). The 15N-urea formed from [5-15N]glutamine must have been derived from glutamine metabolism via PDG, and subsequent incorporation of  into CP via the CPS-I reaction (4, 17) to form Um+1. In addition,
into CP via the CPS-I reaction (4, 17) to form Um+1. In addition,  may appear as [15N]glutamate via reductive amination of α-ketoglutarate by the GDH reaction (4, 17, 18, 29). [15N]Glutamate may then be transaminated to [15N]aspartate followed by transfer of 15N to argininosuccinate and production of Um+2 (16, 17). Previous studies have shown that flux through PDG is up-regulated by cAMP (30) as well as AGM (4). Thus, the questions that arise are as follows. (i) What is the relationship between the elevated [cAMP] flux through PDG and urea synthesis in perfusions with IBMX or IBMX + AGM? (ii) Does the increased urea output in perfusions with IBMX + AGM couple with increased flux through PDG? To address these questions we calculated the flux through PDG by the sum of 15N-labeled products from [5-15N]glutamine between 40 and 60 min of the perfusion as described (4, 18). The calculations show that the rates of PDG are 331 ± 28, 469 ± 78, and 621 ± 61 nmol·min–1·g–1 (means ± S.D.), in control, in perfusion with IBMX, and in perfusions with IBMX + AGM, respectively. These rates are significantly (p < 0.005) higher in IBMX or IBMX + AGM compared with control, and significantly higher in perfusions with IBMX + AGM compared with perfusions with IBMX. These data indicate that IBMX stimulated flux through PDG and that AGM had an additive stimulatory effect. These changes in flux through PDG are tightly linked with the levels of cAMP (Table 1). However, the rates of PDG do not correlate with the rates of total urea output (Fig. 1). These findings indicate that the IBMX-induced decrease in total urea output and the reversal of this decrease by supplementation of AGM (Fig. 1) are independent of the flux through PDG. Furthermore, because physiologic concentrations of [5-15N]glutamine and -ammonia were included in the perfusate (18), and because levels of NAG in freeze-clamped liver extracts were in the control range (5, 18, 19) and did not change following perfusion with or without IBMX or IBMX + AGM (Table 1), the decrease in total urea output should be a consequence of direct inhibition of the urea cycle by IBMX. This conclusion is also supported by the significant fall of [citrulline] in the liver extract (Table 1). IBMX may directly inhibit mitochondrial synthesis of citrulline via either the OTC and/or CPS-I reaction. However, infusion of AGM together with IBMX negated the IBMX-induced inhibition of ureagenesis.
may appear as [15N]glutamate via reductive amination of α-ketoglutarate by the GDH reaction (4, 17, 18, 29). [15N]Glutamate may then be transaminated to [15N]aspartate followed by transfer of 15N to argininosuccinate and production of Um+2 (16, 17). Previous studies have shown that flux through PDG is up-regulated by cAMP (30) as well as AGM (4). Thus, the questions that arise are as follows. (i) What is the relationship between the elevated [cAMP] flux through PDG and urea synthesis in perfusions with IBMX or IBMX + AGM? (ii) Does the increased urea output in perfusions with IBMX + AGM couple with increased flux through PDG? To address these questions we calculated the flux through PDG by the sum of 15N-labeled products from [5-15N]glutamine between 40 and 60 min of the perfusion as described (4, 18). The calculations show that the rates of PDG are 331 ± 28, 469 ± 78, and 621 ± 61 nmol·min–1·g–1 (means ± S.D.), in control, in perfusion with IBMX, and in perfusions with IBMX + AGM, respectively. These rates are significantly (p < 0.005) higher in IBMX or IBMX + AGM compared with control, and significantly higher in perfusions with IBMX + AGM compared with perfusions with IBMX. These data indicate that IBMX stimulated flux through PDG and that AGM had an additive stimulatory effect. These changes in flux through PDG are tightly linked with the levels of cAMP (Table 1). However, the rates of PDG do not correlate with the rates of total urea output (Fig. 1). These findings indicate that the IBMX-induced decrease in total urea output and the reversal of this decrease by supplementation of AGM (Fig. 1) are independent of the flux through PDG. Furthermore, because physiologic concentrations of [5-15N]glutamine and -ammonia were included in the perfusate (18), and because levels of NAG in freeze-clamped liver extracts were in the control range (5, 18, 19) and did not change following perfusion with or without IBMX or IBMX + AGM (Table 1), the decrease in total urea output should be a consequence of direct inhibition of the urea cycle by IBMX. This conclusion is also supported by the significant fall of [citrulline] in the liver extract (Table 1). IBMX may directly inhibit mitochondrial synthesis of citrulline via either the OTC and/or CPS-I reaction. However, infusion of AGM together with IBMX negated the IBMX-induced inhibition of ureagenesis.
The Action of IBMX on Mitochondrial Activity of OTC or CPS-I—To further explore the hypothesis that IBMX may directly inhibit mitochondrial CPS-I and/or OTC activity, experiments were carried out with either isolated mitochondria or freeze-thawed broken mitochondria obtained from the liver of overnight fasted rats. Data in Fig. 2A indicate that IBMX has no effect on the OTC reaction when broken mitochondria were incubated with saturating concentrations of carbamoyl phosphate, ammonia, and ATP. Similar results were obtained with ornithine transcarbamoylase obtained from Sigma (data not shown). Therefore, the inhibitory effect of IBMX may occur at the matrix CPS-I reaction. To examine this possibility, isolated mitochondria were incubated with 15NH4Cl, ATP, ornithine, and increasing concentrations of IBMX. Fig. 2B demonstrates that IBMX inhibited the synthesis of 15N-labeled citrulline in a dose-dependent manner, with an EC50 between 0.6 and 0.9 mm. Because the synthesis of citrulline must reflect carbamoyl phosphate synthesis, and because optimal amounts of ATP and substrates were added to the incubation medium (8–12), the data in Fig. 2B indicate that IBMX directly inhibits CPS-I. In addition, measurement of [NAG] at the end of the incubation showed no significant differences, a finding consistent with that of the liver perfusion studies (Table 2). NAG concentration was 200–300 pmol/mg protein, sufficient to activate CPS-I (5, 8–12, 19).
FIGURE 2.

The action of IBMX on the activity of OTC or CPS-I. Experiments were carried out with isolated mitochondria as outlined under “Experimental Procedures.” A represents the activity of OTC in mitochondrial extract. Incubation was carried out with basic incubation medium, pH 8.5 (see “Experimental Procedures”), supplemented with the following (in mm): 5 ATP, 5 carbamoyl phosphate, and increasing concentrations of IBMX. The reaction was initiated by addition of 5 mm ornithine and stopped after 10 min with the addition of perchloric acid. B represents the percent inhibition of [15N]citrulline synthesis (as proxy for CPS-I activity) with increasing concentrations of IBMX. Isolated mitochondria (≈2 mg of protein·ml–1) were incubated for 20 min with basic medium containing the following (in mm): 5 ATP, 5 ornithine, 5 [5-15N]glutamine, 1 15NH4Cl, 5 pyruvate, and increasing concentrations of IBMX expressed as log [IBMX]. The EC50 (0.6–0.9 mm) was calculated by fitting the data to a sigmoidal curve. C represents the production of total or [15N]citrulline with increasing concentrations of IBMX. Isolated mitochondria (≈2 mg of protein·ml–1) were first loaded with carbamoyl phosphate by preincubation for 5 min with basic medium containing the following (in mm): 2 carbamoyl phosphate, 5 ATP, 5 ornithine, and 5 pyruvate. After 5 min of preincubation, 5 mm [2-15N]glutamine and 1 mm 15NH4Cl were added, and incubation was continued for an additional 10 min. [2-15N]Glutamine was used to follow the synthesis and concentration of NAG at various [IBMX]. The formation of [15N]citrulline was taken as a proxy for 15N-labeled carbamoyl phosphate synthesis from  and bicarbonate. Incubation was stopped by the addition of perchloric acid. Bars are means ± S.D. of 4–6 independent experiments.
and bicarbonate. Incubation was stopped by the addition of perchloric acid. Bars are means ± S.D. of 4–6 independent experiments.
TABLE 2.
Effect of IBMX on mitochondrial oxidative phosphorylation
Isolated mitochondria were obtained from the liver of overnight fasted rats. Mitochondria were added to the polarographic cell containing basic medium without (Control) or with 1 mm IBMX and either substrate at the indicated concentration. After recording state 2 respiration (V2), state 3 respiration (V3, ADP-stimulated) was determined following the addition of 0.3 mm ADP. When most ADP was converted to ATP, the state 4 (V4, ADP-limited) respiration rate was recorded. In a separate measurement, the uncoupled respiration was determined after addition of 10 mm ClCCP. The respiratory rates are represented by oxygen consumption rates given in nanoatom O·mg of protein–1·min–1, and are the means ± S.D. of 3–4 measurements obtained from independent and separate mitochondrial preparations. Results for experiments with substrates of C-II, C-IV and ClCCP are means of two independent measurements. Glu is glutamate.
|
Substrate |
Control |
IBMX |
||||
|---|---|---|---|---|---|---|
| V2 | V3 | V4 | V2 | V3 | V4 | |
| I. Substrates of complex-I | ||||||
| Pyruvate (5 mm) | 8.3 ± 0.1 | 30 ± 1.5 | 19 ± 3.5 | 7.7 ± 1.4 | 21 ± 2.7a | 16 ± 7.56 |
| Malate + Glu (5 mm each) | 14 ± 5.8 | 73 ± 3.1 | 23 ± 3.7 | 12 ± 3.1 | 46 ± 2.7a | 16 ± 1.8a |
| Malate + Glu + ClCCP |
NAb |
54 ± 5.2 |
47 ± 3.6 |
NA |
36 ± 4.1a |
36.6 ± 4.3a |
| II. Substrates of Complex-II | ||||||
| Succinate (5 mm) | 21 ± 8.1 | 84 ± 8.6 | 26 ± 6.1 | 26 ± 7.0 | 72 ± 14 | 26 ± 6.1 |
| Succinate + ClCCP |
NA |
81 |
102 |
NA |
79 |
103 |
| III. Substrates of Complex-IV | ||||||
| TMPD (0.24 mm)/ascorbate (7.2 mm) | 91 ± 13 | 146 ± 27 | 63 ± 22 | 93 ± 16 | 136 ± 32 | 64 ± 24 |
| TMPD/ascorbate + ClCCP | NA | 146 | 211 | NA | 133 | 193 |
p < 0.05 compared with control measurements.
NA, not applicable.
To further characterize the inhibitory action of IBMX on CPS-I, a separate series of experiments was carried out in isolated mitochondria preincubated and loaded with carbamoyl phosphate and NAG for 5 min, after which 15N-labeled ammonia and -glutamine were added, and incubation was continued for an additional 10 min. Data in Fig. 2C demonstrate that the production of total citrulline was constant with an increasing concentration of IBMX, whereas the production of 15N-labeled citrulline from 15N-labeled ammonia was significantly decreased. Under these experimental conditions, total citrulline (determined by HPLC) was synthesized from carbamoyl phosphate and ornithine via the OTC reaction. However, 15N-labeled citrulline (determined by GC-MS as detailed under “Experimental Procedures”) was synthesized from  and
and 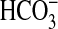 via the CPS-I reaction. This finding provides strong evidence that IBMX directly inhibited CPS-I in a dose-dependent manner. This inhibition is not because of inhibition of NAG synthesis or depletion of the mitochondrial NAG pool.
via the CPS-I reaction. This finding provides strong evidence that IBMX directly inhibited CPS-I in a dose-dependent manner. This inhibition is not because of inhibition of NAG synthesis or depletion of the mitochondrial NAG pool.
The Action of IBMX on Mitochondrial Oxidative Phosphorylation—Because IBMX decreased energetic capacity and oxygen consumption in a liver perfusion system (Table 1), the question arises whether the IBMX-induced inhibition of CPS-I is coupled with inhibition of mitochondrial OXPHOS. We therefore measured mitochondrial respiration with substrates of complex I, complex II, or complex IV. The data in Table 2 demonstrate that IBMX inhibited state 3 respiration with substrates of complex I, including either pyruvate or malate plus glutamate. However, IBMX had no effect on mitochondrial respiration with succinate, a substrate for C-II or TMPD/ascorbate, substrates for C-IV. TMPD donates electrons to the respiratory chain via cytochrome c, which damaged mitochondria easily lose (31). The similarly high respiration rates with TMPD/ascorbate, with or without IBMX (Table 2), indicate that the IBMX-induced inhibition of OXPHOS is not a result of damage to the mitochondrial membrane. Furthermore, because respiration and phosphorylation are coupled, decreased respiration in mitochondria treated with IBMX could result from the inhibition of ATP synthetase. However, the addition of the uncoupler ClCCP to mitochondria treated with IBMX did not increase respiration (Table 2), thus ruling out a defect in ATP synthetase as the underlying mechanism for the IBMX-induced decrease of OXPHOS.
The inhibition of C-I may be due to a direct action of IBMX on the transport of substrates across the mitochondrial membrane and/or on the activity of the electron transport chain. Because malate, succinate, pyruvate, or glutamate are shuttled on different mitochondrial membrane transporters (32), and because pyruvate, glutamate, or malate use different dehydrogenases to generate NADH (33, 34), the current findings demonstrate that IBMX inhibited the activity of complex I. In addition, incubation of isolated mitochondria with [1-13C]- or [U-13C]pyruvate indicated little effect of IBMX on flux through PDH or the tricarboxylic acid cycle as determined by the release of 13CO2. A similar observation was obtained with 13C NMR or GC-MS analysis of 13C-labeled glutamate, aspartate, or intermediates of the tricarboxylic acid cycle at the end of perfusion (data not shown). Therefore, the curtailment of OXPHOS is not because of diminished supply of NADH (C-I substrate) but because of a direct action of IBMX on complex I.
DISCUSSION
Phosphodiesterases cleave the 3′,5-cyclic phosphate moiety of cAMP, thereby destroying second messenger activity. At least 11 distinct PDE families have been identified. Their activity is much greater than adenylate cyclase (7, 35, 36). In the past 3 decades, selective inhibitors of PDEs have been widely used clinically to treat patients with a variety of diseases, including heart failure, hypertension, depression, asthma, and male erectile dysfunction (7, 35, 36). IBMX is a nonspecific inhibitor of PDEs that has been extensively used to potentiate the effects of hormones that act via cAMP. In this study, IBMX was used to explore the hypothesis that AGM stimulates ureagenesis via elevation of [cAMP] (5).
As illustrated in Fig. 3, the current data demonstrate that IBMX induced multiple metabolic disturbances in the liver despite a significant increase in cellular [cAMP]. Together, the data indicate that IBMX induced the following: (i) inhibition of complex I and thus mitochondrial respiration (Table 2); (ii) decreasing O2 consumption during liver perfusion; (iii) depletion of phosphorylation potential and overall hepatic energetic capacity; and (iv) inhibition of CPS-I activity and thereby urea synthesis (Figs. 1 and 2). However, the addition of AGM, a biologic product of arginine decarboxylation (3), negated the action of IBMX and restored hepatic metabolic function toward normal including the following: (i) stimulation of oxygen consumption during liver perfusion (Table 1); (ii) elevation of the hepatic phosphorylation potential and overall energetic capacity (Table 1); (iii) prevention of IBMX-induced inhibition of CPS-I (Fig. 2); and (iv) stimulation of ureagenesis (Fig. 1).
FIGURE 3.

Schematic illustration of IBMX action on liver metabolic network and the protection by agmatine. IBMX induced multiple metabolic disturbances including the following: (i) inhibition of oxidative phosphorylation at the site of complex I, and (ii) inhibition of CPS-I activity and thereby urea synthesis. The addition of AGM with IBMX reversed the inhibitory action of IBMX and restored metabolism toward normal including the following: (i) stimulation of mitochondrial oxidative phosphorylation; (ii) increasing oxygen consumption during liver perfusion; and (iii) prevention of IBMX-induced inhibition of CPS-I and urea synthesis. Presumably, AGM protects against binding of IBMX to one or more active sites of CPS-I and thus prevents the IBMX-induced inhibition of ureagenesis. Both IBMX and/or AGM alter urea synthesis independent of the flux through PDG. The latter was stimulated by either IBMX or AGM and was coupled with elevation of [cAMP]. AGM, which was found to stimulate FAO, may lead to elevated availability of acetyl-CoA for NAG synthesis (5) as well as increased pyruvate carboxylase and thus [oxaloacetate] (14, 15). The latter is used for the synthesis of aspartate, which contributes the second nitrogen for urea synthesis (16, 17). In addition, the stimulation of FAO may increase the availability of FADH2 to complex II (C-II), thereby bypassing the IBMX-induced inhibition of C-I. Together, the current findings and our previous findings (4–6) suggest that AGM is a useful agent to up-regulate ureagenesis in case of hyperammonemia associated with a disturbed urea cycle. Metabolic reactions are as follows: 1, phosphate-dependent glutaminase; 2, carbamoyl-phosphate synthase-I; 3, ornithine transcarbamoylase; 4, glutamate dehydrogenase; 5, aspartate oxaloacetate aminotransferase; 6, pyruvate dehydrogenase; 7, pyruvate carboxylase; 8, fatty acid oxidation; C-I, C-II, C-III, and C-IV indicate respiratory chain complexes; C-V indicates F0F1-ATP synthase; Cyt. c is cytochrome c; ARS indicates argininosuccinate; CACT indicates carnitine:acylcarnitine translocase; 197, indicates up-regulation; 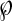 , indicates down-regulation or inhibition by IBMX.
, indicates down-regulation or inhibition by IBMX.
The liver is the major site of conversion of amino-N into urea (1). This process is up-regulated by glucagon via its second messenger, cAMP (2). Therefore, the current findings depict an anomaly in metabolic regulation by demonstrating an inhibition of ureagenesis despite a significant elevation in cAMP concentration. The inhibition of urea synthesis was at the site of CPS-I. Evidence of such inhibition includes the following: (i) the dose-response curves for IBMX inhibition of 15N-labeled citrulline formation in incubation of isolated mitochondria with 15NH4Cl (Fig. 2B); (ii) the inhibition of 15N-labeled citrulline synthesis occurred despite the addition of 5 mm ATP to the incubation medium; and (iii) IBMX had no effect on the synthesis of citrulline when carbamoyl phosphate was added to the incubation medium (Fig. 2C).
CPS-I has multiple active sites as well as “molecular tunnels” that facilitate the passage of reaction intermediates, thereby catalyzing the synthesis of carbamoyl phosphate by four independent chemical reactions (37–39). IBMX-mediated inhibition of CPS-I could reflect one of the following: (i) inhibition of a substrate binding to the enzyme; (ii) disruption of the binding of NAG to its activating site; (iii) the CPS-I reaction requires Mg2+, and IBMX may disrupt the Mg2+-ATP complex, and/or (iv) IBMX may obstruct one or more of the four active sites of CPS-I. Supplementation with AGM completely neutralized the inhibitory action of IBMX on CPS-I activity. AGM was found to elevate NAG synthesis secondary to the stimulation of FAO (5). Because IBMX did not affect mitochondrial [NAG], it is less likely that AGM reversed IBMX-induced inhibition of CPS-I via increased NAG synthesis. Presumably, AGM protects against binding of IBMX to one or more active sites of CPS-I. Precisely how AGM prevents the IBMX-induced inhibition of CPS-I is not obvious and deserves additional studies. One future study may involve examination of structure-function using crystallography (37–39), with the presence or absence of IBMX or IBMX + AGM.
It is also conceivable that the IBMX-induced inhibition of CPS-I is coupled with the IBMX-induced inhibition of complex I (Table 2). Complex-I, the largest complex in mitochondrial respiratory chain (40), participates in the production of a proton gradient across the inner mitochondrial membrane and the transfer of electrons from NADH to ubiquinone, thus providing the proton-motive force used for ATP synthesis (21). Structural investigations suggest that the NADH dehydrogenase moiety may be exposed to the matrix (40), thereby making complex I sensitive to chemicals, drugs, free radicals, and oxidative stress (41). The current observation demonstrates that IBMX inhibits complex I, and this inhibition may initiate a metabolic cascade that exacerbates mitochondrial malfunction. Inhibition of C-I may lead to elevation of superoxide radicals, lipid peroxidation, protein denaturation, and mtDNA mutations (42). It is plausible to suggest that the action of IBMX on CPS-I may be subsequent to IBMX-induced inhibition of complex I activity.
Finally, these data may suggest new experimental applications of IBMX in studies of ureagenesis. An example might be the creation of a model of CPS-I deficiency and the amelioration of such an experimental urea cycle defect by an agent like AGM. In addition, it may be that our findings apply to other PDEs inhibitors, not IBMX alone. This could be a clinically relevant consideration, given the frequency with which various PDEs inhibitors are used clinically in settings such as male erectile dysfunction (35, 36). Therefore, further studies with other inhibitors of PDEs are essential to determine whether the current findings are specific to IBMX or are characteristic of other PDEs inhibitors.
This work was supported, in whole or in part, by National Institutes of Health Grants DK-53761 (to I. N.), DK047870, U54RR019453, and HD26971. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: AGM, agmatine; FAO, fatty acid oxidation; GC-MS, gas chromatography-mass spectrometry; CP, carbamoyl phosphate; CPS-I, carbamoyl-phosphate synthase-I; IBMX, 3-isobutylmethylxanthine; NAG, N-acetylglutamate; OTC, ornithine transcarbamylase; OXPHOS, oxidative phosphorylation; PDE, phosphodiesterase; PDG, phosphate-dependent glutaminase; PDH, pyruvate dehydrogenase; ClCCP, carbonyl cyanide m-chlorophenylhydrazone; HPLC, high pressure liquid chromatography; MSD, mass selective detector.
References
- 1.Meijer, A. J., Lamers, W. H., and Chamuleau, R. A. F. M. (1990) Physiol. Rev. 70 701–744 [DOI] [PubMed] [Google Scholar]
- 2.Exton, J. H., and Park, C. R. (1968) Adv. Enzyme Regul. 6 391–407 [DOI] [PubMed] [Google Scholar]
- 3.Gen, L., Regunathan, D., Barrow, C. J., Esraghi, J., Cooper, R., and Reis, D. J. (1994) Science 263 12231–12234 [DOI] [PubMed] [Google Scholar]
- 4.Nissim, I., Horyn, O., Daikhin, Y., Nissim, I., Lazarow, A., and Yudkoff, M. (2002) Am. J. Physiol. 283 E1123–E1134 [DOI] [PubMed] [Google Scholar]
- 5.Nissim, I., Daikhin, Y., Nissim, I., Luhovyy, B., Horyn, O., Wehhrli, S. L., and Yudkoff, M. (2006) J. Biol. Chem. 281 8486–8496 [DOI] [PubMed] [Google Scholar]
- 6.Nissim, I., Horyn, O., Daikhin, Y., Nissim, I., Luhovyy, B., Phillips, P. C., and Yudkoff, M. (2006) Cancer Res. 66 7824–7831 [DOI] [PubMed] [Google Scholar]
- 7.Dousa, T. P. (1999) Kidney Int. 55 29–62 [DOI] [PubMed] [Google Scholar]
- 8.McGivan, J. D., Bradford, N. M., and Mendes-Mourao, J. (1976) Biochem. J. 154 415–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen, N. S., Cheung, C. W., and Raijman, L. (1980) J. Biol. Chem. 255 10248–10255 [PubMed] [Google Scholar]
- 10.Beliveau, C. G., Cheung, C. W., Cohen, N. S., Brusilow, S., and Raijman, L. (1993) Biochem. J. 292 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawamoto, S., Sonoda, T., Ohtake, A., and Tatibana, M. (1985) Biochem. J. 233 329–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen, N. S., Cheung, C. W., Kyan, F. S., Jones, E. E., and Raijman, L. (1982) J. Biol. Chem. 257 6898–6907 [PubMed] [Google Scholar]
- 13.Fahien, L., Schoder, J. M., Gehred, G. A., and Cohen, P. P. (1964) J. Biol. Chem. 239 1935–1942 [PubMed] [Google Scholar]
- 14.Williams, J. R., Browning, E. T., and Olson, M. S. (1968) Adv. Enzyme Regul. 6 67–100 [DOI] [PubMed] [Google Scholar]
- 15.McGarry, J. D., and Foster, D. W. (1971) J. Biol. Chem. 246 1149–1159 [PubMed] [Google Scholar]
- 16.Nissim, I., Horyn, O., Luhovyy, B., Lazarow, A., Daikhin, Y., Nissim, I., and Yudkoff, M. (2003) Biochem. J. 376 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brosnan, J. T., Brosnan, M. E., Yudkoff, M., Nissim, I., Daikhin, Y., Lazarow, A., Horyn, O., and Nissim, I. (2001) J. Biol. Chem. 276 31876–31882 [DOI] [PubMed] [Google Scholar]
- 18.Nissim, I., Brosnan, M. E., Yudkoff, M., Nissim, I., and Brosnan, J. T. (1999) J. Biol. Chem. 274 28958–28965 [DOI] [PubMed] [Google Scholar]
- 19.Nissim, I., Luhovyy, B., Horyn, O., Daikhin, Y., Nissim, I., and Yudkoff, M. (2005) J. Biol. Chem. 280 17715–17724 [DOI] [PubMed] [Google Scholar]
- 20.Beylot, M., Soloviev, M. V., David, F., Landau, B. R., and Brunengraber, H. (1995) J. Biol. Chem. 270 1509–1514 [DOI] [PubMed] [Google Scholar]
- 21.Ernst-Bernhard, K., Margaret, M. S., Morgan, P. G., and Hoppel, C. L. (2004) J. Biol. Chem. 279 54479–54486 [DOI] [PubMed] [Google Scholar]
- 22.Wehrli, S. L., Reynolds, R., Chen, J., Yager, C., and Segal, S. (2001) NMR Biomed. 14 192–198 [DOI] [PubMed] [Google Scholar]
- 23.Nissim, I., and Weinberg, J. M. (1996) Kidney Int. 49 684–951 [DOI] [PubMed] [Google Scholar]
- 24.Jones, B. N., and Gilligan, J. P. (1983) J. Chromatogr. 266 471–482 [DOI] [PubMed] [Google Scholar]
- 25.Jaworek, D., and Welsch, J. (1985) in Methods of Enzymatic Analysis (Bergmeyer, H. U., ed) 7th Ed., pp. 340–346, Academic Press, New York
- 26.Jaworek, D., Gruber, W., and Bergmeyer, H. U. (1974) in Methods of Enzymatic Analysis (Bergmeyer, H. U., ed) 2nd Ed., pp. 2127–2131, Academic Press, New York
- 27.Pradelles, P., Graassi, J., and Chabandes, D. (1989) Anal. Chem. 61 447–452 [DOI] [PubMed] [Google Scholar]
- 28.Brosnan, T. J., Brosnan, M. E., Charron, R., and Nissim, I. (1996) J. Biol. Chem. 271 16199–16207 [DOI] [PubMed] [Google Scholar]
- 29.Burgess, S. C., Iizuka, K., Jeoung, N. H., Harris, R. A., Kashiwaya, Y., Veech, R. L., Kitazume, T., and Uyeda, K. (2008) J. Biol. Chem. 283 1670–1678 [DOI] [PubMed] [Google Scholar]
- 30.Brosnan, T. J., Ewart, H. S., and Squires, S. A (1995) Adv. Enzyme Regul. 35 131–146 [DOI] [PubMed] [Google Scholar]
- 31.Weinberg, J. M., Venkatachalam, M. A., Roeser, N. F., Suikumar, P., Dong, Z., Senter, R. A., and Nissim, I. (2000) Am. J. Physiol. 279 F927–F943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fiermonte, G., Dolce, V., Arrigoni, R., Runswick, M. J., Walker, J. E., and Palmieri, L. (1999) Biochem. J. 344 953–960 [PMC free article] [PubMed] [Google Scholar]
- 33.Williamson, D. H., Lund, P., and Krebs, H. A. (1967) Biochem. J. 103 514–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krebs, H. A., and Veech, R. L. (1969) Adv. Enzyme Regul. 7 397–413 [DOI] [PubMed] [Google Scholar]
- 35.Bender, A. T., and Beavo, J. A. (2006) Pharmacol. Rev. 58 488–520 [DOI] [PubMed] [Google Scholar]
- 36.Conti, M., and Beavo, J. (2007) Annu. Rev. Biochem. 76 481–511 [DOI] [PubMed] [Google Scholar]
- 37.Thoden, J. B., Huang, X., Kim, J., Raushel, F. M., and Holden, H. M. (2004) Protein Sci. 13 2398–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kothe, M., Eroglu, B., Mazza, H., Samudera, H., and Powers-Lee, S. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 12348–12353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saeed-Kothe, A., and Powers-Lee, S. G. (2002) J. Biol. Chem. 277 7231–7238 [DOI] [PubMed] [Google Scholar]
- 40.Grigorieff, N. (1999) Curr. Opin. Struct. Biol. 9 476–483 [DOI] [PubMed] [Google Scholar]
- 41.Vera, A.-V. (2005) Antioxid. Redox. Signal. 7 1140–1149 [DOI] [PubMed] [Google Scholar]
- 42.Orrenius, S. (2007) Drug Metab. Rev. 39 443–455 [DOI] [PubMed] [Google Scholar]