

Abstract
Expansion of the brain is one of the defining characteristics of modern humans. Seckel syndrome (MIM210600), a disorder of markedly reduced brain and body size1,2, is associated with defective ATR-dependant DNA damage signalling3. Previously, only a single hypomorphic mutation of ATR has been identified in this genetically heterogeneous condition4. We now report that mutations in pericentrin (PCNT) also cause Seckel syndrome, resulting in its loss from the centrosome, where it has key functions anchoring both structural and regulatory proteins5,6. Furthermore, we find that PCNT-Seckel patient cells have defects in ATR-dependent checkpoint signalling, providing the first evidence linking a structural centrosomal protein with DNA damage signalling. These findings also suggest that other known microcephaly genes implicated in either DNA repair responses7 or centrosomal function8,9, may act in common developmental pathways determining human brain and body size.
Seckel syndrome is an autosomal recessive disorder of intra-uterine growth retardation, severe proportionate short stature and marked microcephaly1,2. The ataxia-telancgiectasia and Rad3-related (ATR) gene is mutated in Seckel syndrome (ATR-Seckel)4, and encodes a phosphotidylinositol-3-kinase-like kinase which has distinct, but overlapping functions with ATM, in co-ordinating the response to DNA damage10. ATR is activated by single stranded DNA whilst ATM responds to DNA double strand breaks. MCPH1, encoded by another gene whose mutations cause severe (primary) microcephaly, also acts in the ATR damage response pathway7. Furthermore, other, non-ATR Seckel patient cell lines also exhibit ATR-pathway dysfunction3, suggesting that mutation of further genes encoding proteins involved in this DNA damage response cascade may cause this disorder.
To identify such components, we performed a SNP-microarray genome-wide homozygosity scan on two consanguineous families from the Middle-East with a clinical diagnosis of Seckel syndrome (Supplementary Fig. 1 and 2) and showing cellular evidence of defective ATR signalling. Multipoint linkage analysis identified a novel locus, Sckl4, with a maximum LOD score of 4.03 at 70.9cM on chromosome 21q22.3 between rs1598206 and rs2330591 (Fig. 1 and Supplementary Fig. 2). 55 Refseq annotated genes lay within this interval, including pericentrin (PCNT), which encodes a centrosomal protein. As several centrosomal genes, CenpJ, ASPM and CDK5RAP2, cause the related condition, primary microcephaly8,9, we postulated that PCNT might be the causative gene and therefore sequenced its 47 coding exons in affected individuals from both families.
Figure 1. Schematic of the Sckl4 critical region and the PCNT gene depicting location of identified mutations.
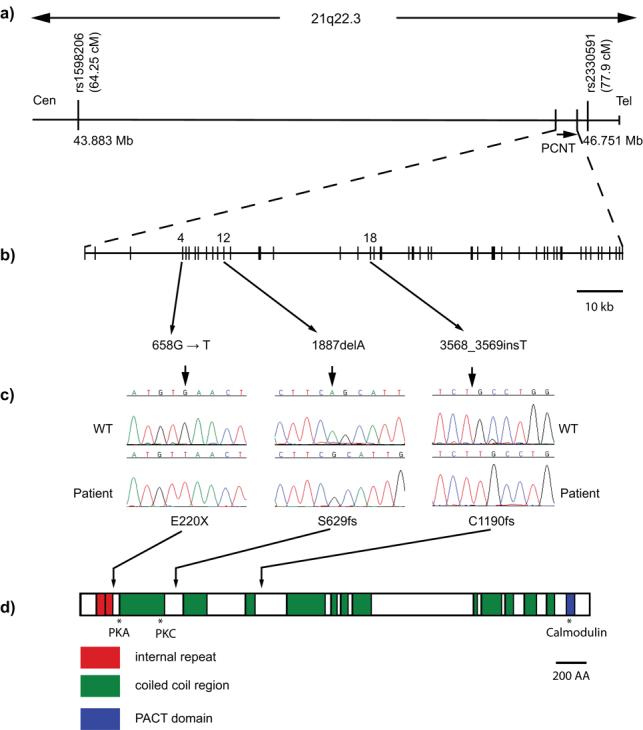
(a) Genetic map of chromosome 21q22.3 with the refined Sckl4 locus, defined by overlapping homozygous chromosomal segments in two consanguineous families, and extending over a 2.9 Mbp interval between SNP markers rs1598206 and rs2330591 (43,883,204 – 46,751,852, UCSC Browser, March 2006 Assembly). Genetic distances, Decode genetic map. (b) PCNT spans 122kb of genomic sequence in 47 exons and encodes a 3336 amino acid protein. (c) Sequence electropherograms of PCNT mutations. (d) Schematic of the pericentrin protein indicating position of identified mutations and protein structure. Pericentrin contains internal repeats (red), coiled-coil regions (green), and a PACT domain (blue). Protein structural regions as predicted by SMART. Sites of interaction with PKA, PKC βII and calmodulin indicated by asterisks.
We identified a homozygous nonsense mutation in exon 4 of family 1 (E220X). A homozygous single base-pair deletion in exon 12 in family 2 (S629fs, Fig. 1c,d) produces a frameshift, predicted to result in premature protein truncation after an additional 65 amino acids. Both mutations segregated with the disease in each family, with all parents being heterozygous for the respective mutations. From screening of additional cases we identified a further patient with a homozygous single base-pair insertion in exon 18, resulting in a frameshift at codon 1190 (C1190fs). Neither this, nor the other two mutations were present in over 200 control alleles screened. Collectively, these mutations disrupt all described mammalian protein isoforms of PCNT11,12. We therefore concluded that homozygous truncating mutations in PCNT cause Seckel syndrome.
Pericentrin (also known as kendrin) is a huge 360kDa coiled-coil protein with a C-terminal PACT domain that targets it to the centrosome (Fig. 1d). Two protein isoforms have been described in humans11, the full length protein, pericentrin B, and a C-terminally truncated isoform, pericentrin A. Pericentrin localises to the pericentriolar material (PCM), where it interacts with several structural centrosomal proteins, including γ-tubulin and PCM113,14 and is important for microtubular nucleation and spindle organisation15,16 Anti-pericentrin antibodies disrupt mitosis, suggesting it is essential for mitotic progression15. However, its Drosophila homolog, D-PLP, though required for timely recruitment of PCM components, is apparently dispensible for cell division17. Furthermore, as well as its structural roles, pericentrin acts as a scaffold to recruit signalling proteins, such as Protein Kinase A (PKA) and Protein Kinase CβII, to the centrosome5,18.
As all mutations identified were homozygous and significantly truncated the protein, this would suggest that they are functionally ‘null’ alleles. To test this prediction, we examined pericentrin localisation in patient lymphoblastoid cell lines (LCLs). Centrosomal pericentrin staining was lost in three independent LCLs, in contrast to control cell lines (Fig. 2a and Supplementary Fig. 3), both in interphase and mitosis.
Figure 2. Pericentrin localisation and function is disrupted in PCNT-Seckel cell lines.
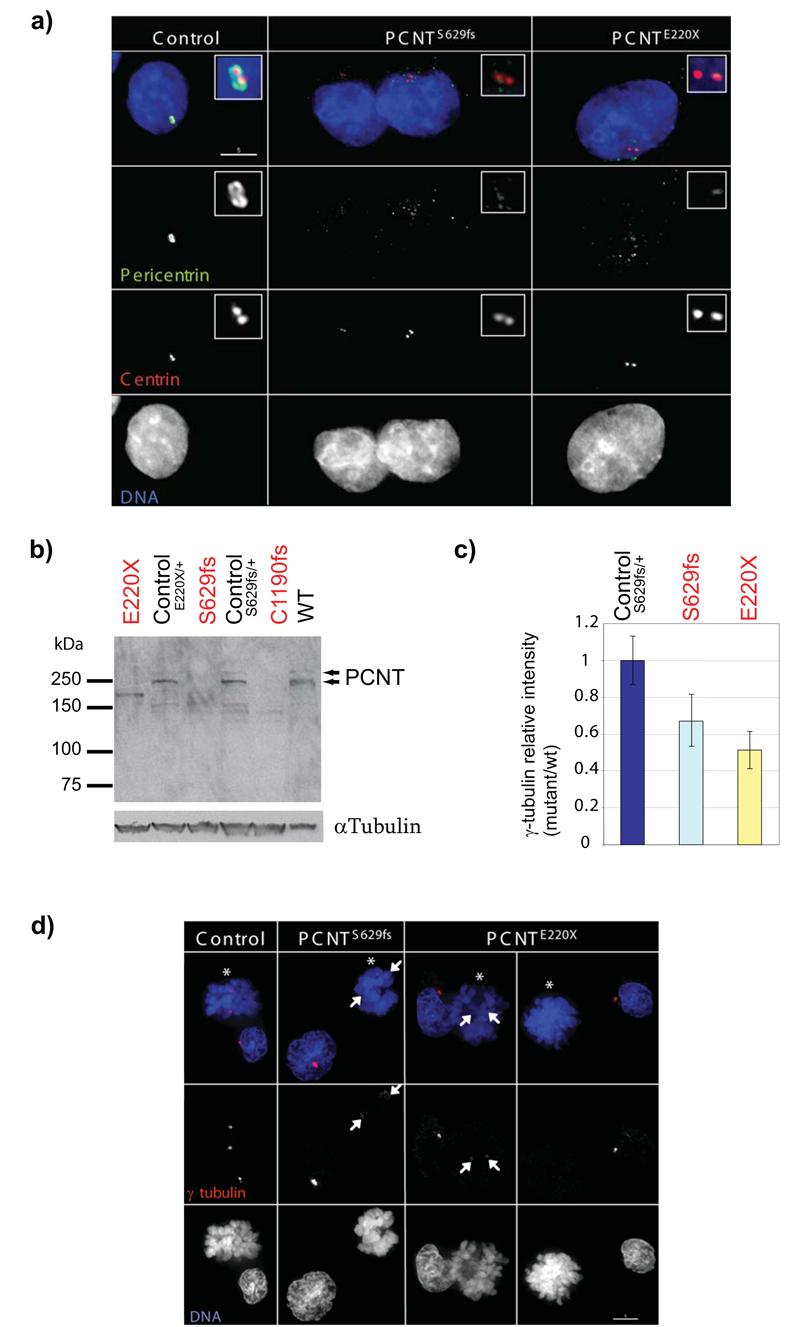
(a) Pericentrin is not localised to the pericentiolar material in PCNT-Seckel cells. Deconvolved immunofluorescent images from PCNT-Seckel and control lymphoblastoid cell lines. Pericentrin (ab448-100, green) and centrin (red). Scale bar 5μm. Control, heterozygote relative, PCNT220X/+. (b) Immunoblot of LCL cell lysates with Pericentrin ab448-100 antibody, that detects both Pericentrin A and B isoforms. Two pericentrin (arrowheads) isoforms are absent from PCNT-Seckel cells, but present in control lymphoblastoid cells from heterozygous relatives. A smaller protein product of ∼170kD is detected in the PCNT220X cell line,that might represent an aberrant truncated PCNT protein product. Loading control, alpha-tubulin. (c,d) γ–tubulin localisation is frequently reduced or absent during mitosis in PCNT-Seckel cells. (c) Quantification of γ-tubulin signal in PCNT-Seckel mitotic cells at prometaphase and metaphase, relative to wild-type. n=20. error bar, s.d. γ-tubulin signal intensity is significantly reduced relative to wild type cells (p<0.05, S629fs; p<0.001, E220X) (d) In PCNT-Seckel mitotic cells (astericks), γ–tubulin centrosomal staining is reduced (arrows) or absent, relative to centrosomal staining in adjacent interphase cells, or in control cells from a heterozygous relative.
We also examined protein expression by immunoblotting (Fig. 2b). Two bands were detected in controls (Wild-type (WT), and heterozygous relatives, PCNTE220X/+, PCNTS629fs/+), representing two isoforms of pericentrin, which were not present in mutant LCLs. A smaller ∼170kD band was present in PCNTE220X lymphoblastoid cells, (potentially representing an aberrant truncated PCNT protein product), while no significant PCNT protein was apparent in the other two mutant cell lines, therefore supporting the molecular genetic findings that protein function is significantly, if not completely, disrupted.
We next examined other components of the centrosome, to establish if they were affected by the absence of pericentrin. ASPM, PCM-1, ninein and centrin were all localised normally (Fig 2a and Supplementary Fig. 4). However, during mitosis, γ-tubulin was significantly reduced or absent at prometaphase/metaphase (Fig 2c, d), consistent with previous RNAi experiments6.
Other patients with Seckel syndrome are defective in the ATR DNA damage response pathway3. We therefore assessed PCNT-Seckel patient cell lines to see if loss of this key centrosomal protein is associated with the defects in ATR signalling previously described for this disorder3,4. Indeed, PCNT-Seckel (PCNTC1190fs, PCNTE220X and PCNTS629fs) LCLs exhibited defective UV-induced G2-M checkpoint arrest, similar to that seen in ATR-Seckel patients (Fig. 3a), in contrast to cell lines established from heterozygote relatives (PCNTE220X/+, PCNTS629fs/+) and an unrelated wild type control. This suggested that mutations in PCNT impact on ATR-dependent cell cycle checkpoint activation. Similarly, PCNT-Seckel LCLs also exhibited elevated nuclear fragmentation following replication fork stalling (Fig. 3b) and supernumerary mitotic ‘centrosomes’ after prolonged mitotic arrest with nocodazole (Fig 3c), two additional cellular phenotypes caused by impaired ATR-signalling3.
Figure 3. PCNT is required for ATR dependent DNA damage signalling.
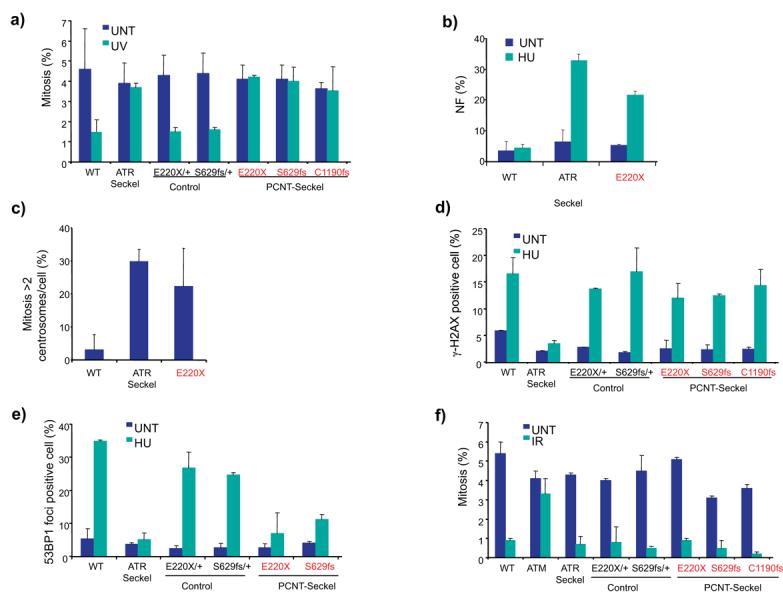
(a) G2-M checkpoint arrest was observed 2 h post UV treatment in control but not in ATR- or PCNT-Seckel LCLs. The mitotic index was examined 2 hours after treatment with 5 J m−2 UV. Average of 3 experiments, error bars, s.d. Controls:- heterozygote relatives PCNTE220X/+ and PCNTS629fs/+ ; and an unrelated wild type (WT) control. UNT, untreated; UV, UV-C treated. Mitotic index is significantly increased in PCNT-Seckel cells compared with Wild-Type cells after UV treatment (p<0.001, E220X; p<0.01, S629fs; p<0.05, C1190fs) (b) ATR- and PCNT-Seckel LCL cells have increased hydroxyurea-induced nuclear fragmentation. Nuclear fragmentation was examined 24 h after treatment with 5mM hydroxyurea (HU). Percentage of cells displaying nuclear fragmentation (NF) is shown (p<0.001 WT versus PCNT-Seckel E220X) (c) ATR- and PCNT-Seckel LCL cells exhibit elevated levels of supernumerary centrosomes in mitosis. Percentage of mitotic (phosphohistoneH3 (Ser10) positive cells) with > 2 γ-tubulin stained foci (centrosomes) determined following 24 h incubation with nocodazole (p<0.05 WT versus E220X) (d) γH2AX foci formation is normal in PCNT-Seckel LCLs. Phosphorylation of the histone H2AX (termed γH2AX) by the PI3K-kinases is one of the earliest detectable responses to DNA damage, and is ATR-signalling specific after hydroxyurea treatment. Percentage of γH2AX foci was determined 2 hours after treatment with 5mM hydroxyurea. (e) ATR- and PCNT-Seckel cells show significantly reduced 53BP1 foci formation after treatment with 5mM hydroxyurea for 2 hrs, reflecting impairment in ATR/Chk1 dependent signalling. Immunofluorescent staining with anti-BrdU confirmed that all LCLs had equivalent S-phase populations (range: 20-23%) prior to exposure to HU (p<0.005 E220X; and p<0.001, S629fs versus WT after HU). (f) G2-M checkpoint arrest after Ionizing Radiation (IR) is normal in PCNT-Seckel and ATR-Seckel LCLs but not in Ataxia Telangiectasia mutated (ATM) LCLs.
Since, these findings suggested a direct role for PCNT in ATR-dependent DNA damage response signalling, we examined further steps in this signalling cascade in PCNT-Seckel cells. We found that ATR-dependent γH2AX formation was normal in Seckel-PCNT LCLs, similar to controls (Fig 3d). This was distinct from ATR-Seckel LCLs and suggested that ATR is able to phosphorylate a key substrate in PCNT-Seckel syndrome. Thus, Pericentrin functions downstream of ATR kinase activation in this pathway.
We next examined hydroxyurea-induced 53BP1 foci formation in PCNT-Seckel cell lines. Strikingly, both PCNT-Seckel and ATR-Seckel LCLs failed to form such foci in contrast to control cell lines (Fig 3e). This phenotype is dependent on both ATR and Chk1 kinases19, indicating that PCNT has a downstream role in ATR-pathway function.
To determine whether PCNT mutations affect other DNA damage responses, we assessed the G2-M checkpoint response to DNA double strand breaks induced by ionising radiation. This response is specifically dependent on ATM and not ATR signalling. PCNT-Seckel LCLs arrested as efficiently as control LCLs under these conditions. ATR-Seckel, unlike ATM mutant LCLs, also had a normal response, confirming the ATR-independence of this assay (Fig 3f). Therefore, we concluded that mutations in PCNT have specific effects on the downstream stage of the ATR-pathway including G2-M checkpoint activation.
We also performed siRNA experiments in HeLa cells to provide confirmatory evidence that PCNT is required for ATR-dependent signalling. siRNA depletion of PCNT also results in defective ATR-dependent (UV) checkpoint activation (Supplementary Fig. 5), as we had observed in PCNT Seckel LCLs. Similarly, no defect was seen in ATM-dependant checkpoint response after ionizing radiation. This substantiates our findings that PCNT mutations cause imparied ATR signalling.
PCNT is a key structural centrosomal protein nucleating microtubular assembly during mitosis15,16. It was thus surprising to identify homozygous truncating mutations that appear to cause absence of known forms of the PCNT protein from centrosomes. So remarkably, PCNT would seem to be dispensable for most aspects of human development. Pericentrin mutations do however dramatically reduce both body and brain size by ∼ 8 standard deviations below the norm. Other centrosomal genes (ASPM, CDK5RAP2, CENPJ) have been implicated in determining human brain size, being mutated in primary microcephaly. They are presumed to act through effects on neurogenic mitosis8,9,20, perhaps modulating symmetric/asymmetric cell division. Given PCNT's role in mitotic spindle assembly15,16, perturbation of cell division may similarly result in globally reduced cell number, so additionally causing reduced body size. However for ATR, MCPH1 and now PCNT genes, there is a specific correlation between defects in ATR signalling and reduced brain and body size4,7. This suggests an alternative explanation, that impaired ATR signalling might itself, directly cause these phenotypes through impaired mitotic progression or reduced cell survival.
Primary microcephaly genes have undergone significant adaptive evolution in primates, suggesting that functional alterations in the proteins they encode may have contributed to the evolution of human brain size21. Morphologically, Seckel syndrome also has evolutionary parallels. Homo Floresiensis, the recently described Indonesian hominid, has similarly marked reduced brain and body size (cranial volume -6.1 s.d, height -9.5 s.d, relative to modern humans)22. Consequently, changes in this or similar genes could underlie the anatomical differences seen in this hominid23.
Through study of a human disease phenotype, we have for the first time implicated a structural protein of the centrosome in the ATR-dependent DNA damage response. Centrosomes have a central role in regulating cell cycle progression24 and pericentrin has an established role in recruiting regulatory proteins to the centrosome5,18. Therefore a role in checkpoint signalling would be consistent with its known functions. Components of the ATR-pathway have been observed to localise to the centrosome25, most notably Chk126,27. Chk1 centrosomal localisation, mediated by activating phosphorylation at Serine317 and 345 residues28, is enhanced by DNA damage. At the centrosome, Chk1 is thought to delay G2/M transition through inhibiting activation of Cyclin B-Cdk1 by Cdc25 27. Intriguingly, fusion of Chk1 with a PACT domain prevents G2/M mitotic progression27, suggesting that endogenous pericentrin, one of only two PACT domain containing proteins, could provide the means of Chk1 localisation to mediate G2/M checkpoint arrest (model, Fig. 4). Alternatively, PCNT may not be required for localisation of Chk1, but may be necessary for Chk1 to transmit its signal to downstream centrosomally localized components to effect G2/M arrest. Future investigations based on these possibilities will be valuable in understanding the mechanism of Chk1 relocalisation and activation at the centrosome.
Figure 4. Model of Pericentrin's role in ATR-dependent G2/M checkpoint arrest.
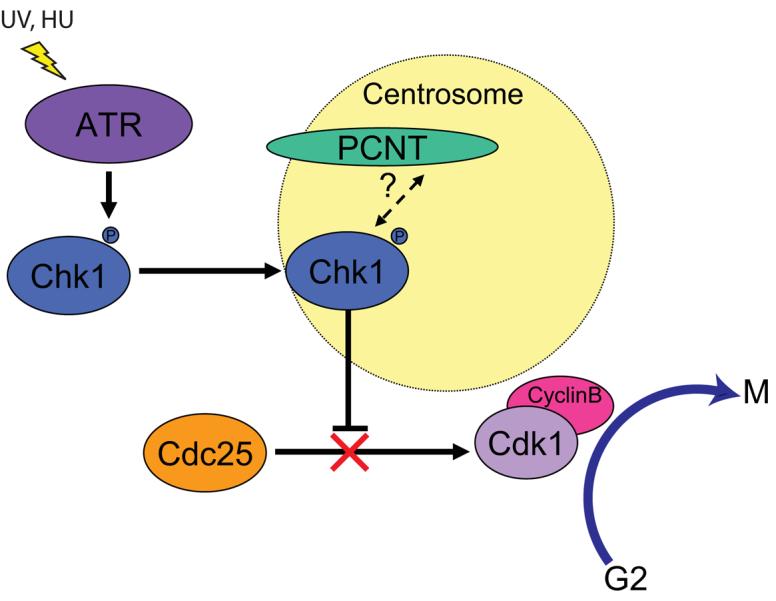
Low-dose UV and Hydroxyurea activate ATR kinase which then phosphorylates downstream targets including Chk1 kinase. Activating phosphorylation is required for accumulation of Chk1 at the centrosome after DNA damage28. Direct or indirect binding of Chk1 by pericentrin could mediate Chk1's localisation to the centrosome. Chk1 localisation at the centrosome inhibits Cdc25, preventing Cyclin B/Cdk1 activation27 and thus the transition from G2 in to mitosis.
The identification of Pericentrin mutations in Seckel syndrome also provides an interesting convergence between microcephaly genes implicated in ATR signalling (MCPH1, ATR) and those involved in centrosomal function (ASPM, CDK5RAP2, CENPJ). This suggests that further biochemical and developmental investigation is likely to be fruitful in delineating common cellular pathways responsible for the brain and body size phenotypes in these syndromes.
Methods
Affected individuals with Seckel syndrome
All affected individuals included in this study had a diagnosis of Seckel syndrome, having intrauterine growth retardation, severe proportionate short stature, marked microcephaly and developmental delay. Blood samples were obtained with informed consent from affected children, parents and unaffected siblings. Lymphoblastoid cell lines were established by EBV transformation and Genomic DNA extracted using standard methods. The study was approved by the Scottish Multicentre Research Ethics Committee (04:MRE00/19), and the University of Sussex School of Life Sciences Research Governance Committee.
Genotyping and linkage analysis
SNP microarray genotyping was performed by AROS Applied Biotechnology (Denmark) using Affymetrix Human Mapping 50k Xba240 Genechips. SNP genotypes were analysed using Alohomora 29 and multipoint linkage analysis performed with GENEHUNTER (version 2.0β) under a model of autosomal recessive inheritance with full penetrance, using a disease allele frequency estimated at 1 in 1,000. Decode genetic map distances and Affymetrix SNP allele frequencies were used.
Mutation detection
Primers were designed using the ExonPrimer tool in UCSC Genome Browser to amplify coding exons of pericentrin (primer sequences listed in Supplementary Table 1). Purified PCR amplification products were sequenced using dye-terminator chemistry and electrophoresed on an ABI 3730 capillary sequencer (Applied Biosystems). Mutation analysis was performed using Mutation Surveyor (Softgenetics). Anonymised control samples were screened by sequencing.
Cell lines and cell culture
LBLs were cultured in RPMI 1640 with 15% fetal calf serum (FCS). Cells used were Wild-type (WT; GM02188), ATR-Seckel (DK0064), Ataxia Telangiectasia-mutated (ATM; GM03189D), all of which are described previously 4,7. PCNTE220X, (CV1559; affected Seckel, PCNT 658G>T), PCNTE220X/+, (CV1584; unaffected brother of CV1559), PCNTS629fs (CV1576; affected Seckel, PCNT 1887delA), PCNTS629fs/+ (CV1582; unaffected mother of CV1576) and PCNTC1190fs (12061; affected Seckel, PCNT 3568_3569insT).
Immunofluorescence and Western Blotting
Immunostaining was performed as previously described30. Antibodies were used at the following dilutions: pericentrin (Abcam, ab448-100) 1:1000; pericentrin (N-20) (Santa Cruz Biotechnology) 1:100; γ-tubulin (Sigma) 1:1000; Centrin (20H5, J. Salisbury) 1: 1000; PCM-1 (A.Merdes) 1: 1000; ASPM (Bethyl labs, IHC-00058); 1:250; Ninein (A.Merdes) 1:100. Secondary antibodies were FITC or Texas Red conjugated (Jackson Immunoresearch lab), 1:200. Three dimensional data sets were collected with a DeltaVision system (Applied Precision, Issaquah, WA) based on an Olympus IX-70 with a Chroma Technology Sedat filter set driven by SoftWorx software under standard conditions. All images were archived as raw (r3d) and deconvolved (d3d) files. Images were subjected to standard deconvolution algorithm. Three dimensional data sets were converted to Quick Projections in SoftWorx and then converted to TIFF files and imported into Adobe Photoshop for final presentation. For the quantification of γ-tubulin staining in mitosis, a defined volume containing the centrosome was used to measure the total intensity of the signal. The same volume was used to identify the background noise as intensity outside the cells and the non-centrosomal gamma tubulin. The ratios of centrosome-bound and dispersed gamma tubulin was averaged for each slide after background subtraction and the relative intensity was calculated in comparison to the WT control slide.
Immunoblotting was performed using pericentrin (Abcam, ab448-100) antibody at 1:1000, and HRP conjugated anti-rabbit IgG secondary 1:5000 (Cell signalling 7074), detected with ECL-Plus (Amersham).
DNA damage response assays
For ATR-dependent G2-M arrest cells were untreated (UNT) or irradiated with 5 J/m2 UV-C (UV) and immediately seeded into complete medium, incubated for 2 hrs, before being cytospun onto poly-D-lysine coated slides and stained with DAPI and anti-phosphoHistone H3 as previously described7. For the ATM-dependent G2-M arrest cells were either untreated (UNT) or irradiated with 2 Gy ionising radiation (IR) and processed as above. For γH2AX formation and 53BP1 foci formation, cells were either untreated (UNT) or treated with 5 mM hydroxyurea (HU) for 2 hrs and processed for immunofluoresence (IF) staining as previously described3. Cells were also pulse labelled (15 mins) with 50μM BrdU to allow quantification of S-phase using anti-BrdU by IF. Supernumerary mitotic centrosomes were determined following treatment with 1.5μM nocodazole for 24 hrs as previously described7. Anti-γH2AX was obtained from Upstate Technology (Hampshire, UK), anti-53BP1 (BL181) from Universal Biologicals (Cambridge, UK), anti-BrdU from Autogen Bioclear (Wiltshire, UK) and anti-γ tubulin from Sigma-Aldrich UK Ltd (Dorset, UK).
RNAi
HeLa cells were transfected with 10nmol of Invitrogen Stealth™ duplex oligoribonucleotides using siPORT™ NeoFX™ Transfection Agent (Ambion, standard protocol). siRNA oligo sequences, Supplementary table 1. 72 hours later G2/M checkpoint assays were performed as outlined above.
Supplementary Material
Acknowledgements
We thank the families and their clinicians for their participation in this study; C. Hayward for contributing control samples; S. McKay and the MRC HGU core sequencing service for advice and technical support; Craig Nicol for help with figure preparation; X. Fant, V. Van Heyningen, and N. Hastie for discussions and comments. W. Fergusson, St Mary's Hopsital, Manchester, for LCL transformation. Andreas Merdes and Jeff Salisbury for kindly sharing antibody reagents. The A.P.J lab is funded by the MRC, the M.O'D. lab is funded by CRUK and the MRC, and the P.A.J laboratory is funded by the MRC, UK LRF, IACR and EU grants (FIGH-CT-200200207) (DNA repair) and FI6R-CT-2003-508842 (RiscRad).. PV/WCE are funded by The Wellcome Trust, of which WCE is a Principal Research Fellow. A.P.J. is an MRC Senior Clinical Fellow and M.O'D. is a CRUK Senior Cancer Research Fellow.
Footnotes
Database Accession Numbers
Pericentrin Transcript, NM_006031.4; protein NP_006022.
URLs
UCSC Human Genome Browser
SMART, (Simple Modular Architecture Research Tool)
References
- 1.Majewski F, et al. Studies of microcephalic primordial dwarfism I: approach to a delineation of the Seckel syndrome. Am J Med Genet. 1982;12:7–21. doi: 10.1002/ajmg.1320120103. [DOI] [PubMed] [Google Scholar]
- 2.Seckel HPG. Bird-headed Dwarfs: Studies in Developmental Anthropology Including Human Proportions. Springfield, Illinois: Charles C Thomas; 1960. [Google Scholar]
- 3.Alderton GK, et al. Seckel syndrome exhibits cellular features demonstrating defects in the ATR-signalling pathway. Hum Mol Genet. 2004;13:3127–38. doi: 10.1093/hmg/ddh335. [DOI] [PubMed] [Google Scholar]
- 4.O'Driscoll M, et al. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 5.Diviani D, et al. Pericentrin anchors protein kinase A at the centrosome through a newly identified RII-binding domain. Curr Biol. 2000;10:417–20. doi: 10.1016/s0960-9822(00)00422-x. [DOI] [PubMed] [Google Scholar]
- 6.Zimmerman WC, et al. Mitosis-specific anchoring of gamma tubulin complexes by pericentrin controls spindle organization and mitotic entry. Mol Biol Cell. 2004;15:3642–57. doi: 10.1091/mbc.E03-11-0796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alderton GK, et al. Regulation of mitotic entry by microcephalin and its overlap with ATR signalling. Nat Cell Biol. 2006;8:725–33. doi: 10.1038/ncb1431. [DOI] [PubMed] [Google Scholar]
- 8.Bond J, et al. ASPM is a major determinant of cerebral cortical size. Nat Genet. 2002;32:316–20. doi: 10.1038/ng995. [DOI] [PubMed] [Google Scholar]
- 9.Bond J, et al. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet. 2005;37:353–5. doi: 10.1038/ng1539. [DOI] [PubMed] [Google Scholar]
- 10.Shiloh Y. ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev. 2001;11:71–7. doi: 10.1016/s0959-437x(00)00159-3. [DOI] [PubMed] [Google Scholar]
- 11.Flory MR, et al. The centrosomal proteins pericentrin and kendrin are encoded by alternatively spliced products of one gene. Genomics. 2003;82:401–5. doi: 10.1016/s0888-7543(03)00119-8. [DOI] [PubMed] [Google Scholar]
- 12.Miyoshi K, et al. Characterization of pericentrin isoforms in vivo. Biochem Biophys Res Commun. 2006;351:745–9. doi: 10.1016/j.bbrc.2006.10.101. [DOI] [PubMed] [Google Scholar]
- 13.Li Q, et al. Kendrin/pericentrin-B, a centrosome protein with homology to pericentrin that complexes with PCM-1. J Cell Sci. 2001;114:797–809. doi: 10.1242/jcs.114.4.797. [DOI] [PubMed] [Google Scholar]
- 14.Dictenberg JB, et al. Pericentrin and gamma-tubulin form a protein complex and are organized into a novel lattice at the centrosome. J Cell Biol. 1998;141:163–74. doi: 10.1083/jcb.141.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doxsey SJ, et al. Pericentrin, a highly conserved centrosome protein involved in microtubule organization. Cell. 1994;76:639–50. doi: 10.1016/0092-8674(94)90504-5. [DOI] [PubMed] [Google Scholar]
- 16.Purohit A, et al. Direct interaction of pericentrin with cytoplasmic dynein light intermediate chain contributes to mitotic spindle organization. J Cell Biol. 1999;147:481–92. doi: 10.1083/jcb.147.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez-Campos M, et al. The Drosophila pericentrin-like protein is essential for cilia/flagella function, but appears to be dispensable for mitosis. J Cell Biol. 2004;165:673–83. doi: 10.1083/jcb.200402130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen D, et al. Centrosomal anchoring of protein kinase C betaII by pericentrin controls microtubule organization, spindle function, and cytokinesis. J Biol Chem. 2004;279:4829–39. doi: 10.1074/jbc.M311196200. [DOI] [PubMed] [Google Scholar]
- 19.Sengupta S, et al. Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest. J Cell Biol. 2004;166:801–13. doi: 10.1083/jcb.200405128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woods CG, et al. Autosomal Recessive Primary Microcephaly (MCPH): A Review of Clinical, Molecular, and Evolutionary Findings. Am J Hum Genet. 2005;76:717–28. doi: 10.1086/429930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ponting C, et al. Evolution of primary microcephaly genes and the enlargement of primate brains. Curr Opin Genet Dev. 2005;15:241–8. doi: 10.1016/j.gde.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 22.Brown P, et al. A new small-bodied hominin from the Late Pleistocene of Flores, Indonesia. Nature. 2004;431:1055–61. doi: 10.1038/nature02999. [DOI] [PubMed] [Google Scholar]
- 23.Martin RD, et al. Flores hominid: new species or microcephalic dwarf? Anat Rec A Discov Mol Cell Evol Biol. 2006;288:1123–45. doi: 10.1002/ar.a.20389. [DOI] [PubMed] [Google Scholar]
- 24.Doxsey S, et al. Centrosomes in cellular regulation. Annu Rev Cell Dev Biol. 2005;21:411–34. doi: 10.1146/annurev.cellbio.21.122303.120418. [DOI] [PubMed] [Google Scholar]
- 25.Zhang S, et al. Centrosomal localization of DNA damage checkpoint proteins. J Cell Biochem. 2007;101:451–65. doi: 10.1002/jcb.21195. [DOI] [PubMed] [Google Scholar]
- 26.Loffler H, et al. DNA Damage-Induced Accumulation of Centrosomal Chk1 Contributes to its Checkpoint Function. Cell Cycle. 2007;6 doi: 10.4161/cc.6.20.4810. [DOI] [PubMed] [Google Scholar]
- 27.Kramer A, et al. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat Cell Biol. 2004;6:884–91. doi: 10.1038/ncb1165. [DOI] [PubMed] [Google Scholar]
- 28.Niida H, et al. Specific role of Chk1 phosphorylations in cell survival and checkpoint activation. Mol Cell Biol. 2007;27:2572–81. doi: 10.1128/MCB.01611-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruschendorf F, et al. ALOHOMORA: a tool for linkage analysis using 10K SNP array data. Bioinformatics. 2005;21:2123–5. doi: 10.1093/bioinformatics/bti264. [DOI] [PubMed] [Google Scholar]
- 30.Dodson H, et al. Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. Embo J. 2004;23:3864–73. doi: 10.1038/sj.emboj.7600393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.