

Abstract
This study presents an interconnected approach for circumventing two inherent limitations associated with studies defining the natural history of influenza A viruses in wild birds. The first limiting factor is the ability to maintain a cold chain from specimen collection to the laboratory when study sites are in more remote locations. The second limiting factor is the ability to identify all influenza A virus HA subtypes present in an original sample. We report a novel method for molecular subtyping of avian influenza A virus hemagglutinin genes using degenerate primers designed to amplify all known hemagglutinin subtypes. It was shown previously that templates larger than 200 bp were not consistently amplifiable from ethanol-fixed cloacal swabs. For this study, new primer sets were designed within these constraints. This method was used to perform subtyping RT-PCR on 191 influenza RNA-positive ethanol-fixed cloacal swabs obtained from 880 wild ducks in central Alaska in 2005. Seven different co-circulating hemagglutinin subtypes were identified in this study set, including H1, H3, H4, H5, H6, H8, and H12. In addition, 16% of original cloacal samples showed evidence of mixed infection, with samples yielding from two-to-five different hemagglutinin subtypes. This study further demonstrates the complex ecobiology of avian influenza A viruses in wild birds.
Keywords: Influeza A virus, Hemagglutinin, Subtype, RT-PCR, Surveillance
Introduction
Influenza A viruses are globally distributed in wild avian species. They have most frequently been recovered from aquatic birds of the orders Anseriformes and Charadriiformes which are thought to represent the major natural reservoirs for influenza A viruses (Krauss et al., 2004; Munster et al., 2007; Slemons et al., 1974; Spackman et al., 2005). At least 105 species of the more than 9000 species of wild birds have been identified as harboring influenza A viruses (Hanson et al., 2003; Munster et al., 2007; Stallknecht and Shane, 1988). This pool of antigenically and genetically diverse low pathogenicity (LP) influenza A viruses maintained in wild birds are commonly referred to as avian influenza viruses (AIV) and include all known influenza A virus hemagglutinin (HA) (H1–H16) and neuraminidase (NA) (N1–N9) subtypes (Fouchier et al., 2005). At least 103 of the possible 144 type A influenza A virus HA-NA combinations have been found in wild birds (Alexander, 2006; Munster et al., 2007). However, in spite of extensive research efforts over many years, it is still unclear how the antigenic and genetic diversity of the AIVs are maintained in their natural wild bird reservoirs.
The current panzootic of Asian-lineage highly pathogenic (HP) H5N1 AIV appears to be unique in the era of modern influenza virology, resulting in the deaths of millions of poultry in 64 countries on three continents either from infection or culling. There are also significant zoonotic implications of this panzootic (Taubenberger, Morens, and Fauci, 2007), with 335 documented cases in humans, resulting in 206 deaths in 12 countries since 2003 (as of November 2007) (WHO, 2007). The Asian lineages of HP H5N1 AIV have also caused symptomatic, even lethal, infections of wild birds in Asia and Europe, and there is evidence suggesting that while poultry may be the current reservoir, migratory wild birds could be involved in the spread of this avian panzootic (Bragstad et al., 2007). Concern is further heightened since wild birds are also likely to be the reservoir of influenza A viruses that switch hosts and stably adapt to mammals including horses, swine, and humans (Webster et al., 1992). Once an H5 or H7 wild bird–origin AIV is established in poultry, the poultry-adapted virus can sporadically and unpredictably evolve into HP AIVs through mutational events resulting in a polybasic amino acid cleavage site within the HA (Alexander, 2006). In addition, the last three human influenza pandemic viruses all contained two or more novel genes that were very similar to those found in wild birds (Kawaoka, Krauss, and Webster, 1989; Scholtissek et al., 1978; Taubenberger et al., 2005). Thus, there is an increased need to perform influenza virus surveillance in wild bird populations.
Two inherent limitations of investigations into understanding of the natural history of influenza A viruses in wild birds are maintaining a strict cold chain for the samples when working in remote locations and then detecting all the HA subtypes of viruses present in the original samples. In viral isolate-based investigations, the logistics and inability to maintain a cold chain following specimen collection and during shipping can contribute to decreased frequencies of virus recovery. Second, detecting mixed influenza A virus infections in wild birds, which appear to be playing a key role in the natural history of these viruses, is extremely time and resource demanding. If multiple strains of AIV are present in the original sample, one strain of virus commonly outgrows others in the embryonating chicken eggs (ECE), an aberrant host system, and the other strains remain undetected. If an AIV isolate is recovered from a sample, subtyping is then determined by HA-and NA-inhibition tests (HIT and NIT) using reference antibodies and antigens (Lee, Senne, and Suarez, 2006). If a dominant strain does not emerge, the HIT and NIT results are inconclusive and the isolate warrants repassaging in ECE followed by dilution, or even limiting dilution. This permits viral adaptation to the aberrant chicken culture system and a dominant viral strain emerges (Slemons et al., 2003). Therefore, over the years, the frequency of mixed influenza A virus infections in wild birds has likely been greatly underestimated. Being able to use ethanol-fixed swabs (or another appropriate preserving agent) for detecting type A influenza viruses and still be able to determine the HA subtypes would at least partially circumvent the cold chain problem while facilitating efforts to gain new knowledge about mixed influenza A virus infections in their natural hosts in new geographic areas.
In previous studies, we demonstrated the utility of RT-PCR based methods for influenza A virus detection in ethanol-fixed human nasal swabs (Krafft et al., 2005) and ethanol-fixed cloacal swabs from wild birds (Evers, Slemons, and Taubenberger, 2007; Runstadler et al., 2007). This work demonstrated that successful RT-PCR from ethanol-fixed samples required templates smaller than 200 bp (Evers, Slemons, and Taubenberger, 2007). Several RT-PCR based methods have been published for HA subtyping, but these require templates too large for fixed-swab surveillance (Bragstad et al., 2005; Hoffmann et al., 2001; Phipps, Essen, and Brown, 2004). A variety of real-time RT-PCR subtyping assays have been described (Ong et al., 2007; Payungporn et al., 2006; Spackman et al., 2002; Stone et al., 2004), but most are specific to HA subtypes, and a simple system to screen and unequivocally determine all HA subtypes by RT-PCR from either fixed or unfixed specimens has not been previously described. Here, we describe an RT-PCR based method that allows HA subtype determination from AIV RNA preserved in ethanol-fixed cloacal swabs and use this method to examine HA subtype diversity from wild duck-origin AIV.
Methods
Sample collection
Clocal swabs from 880 wild ducks were collected at different locations within the Minto Flats State Game Refuge, Alaska from August 8–21, 2005 as previously described (Runstadler et al., 2007). RNA was isolated from these samples and screened by real-time RT-PCR for the influenza A virus RNA matrix gene segment (Runstadler et al., 2007). A total of 225 (25.6%) of the samples were previously identified as RT-PCR positive for influenza A virus RNA (Runstadler et al., 2007).
Primer design
Avian influenza virus HA gene sequences of subtypes H1–H16 from North America and Eurasia were downloaded from the Influenza Virus Resource page of the NCBI and aligned using the Megalign program (Lasergene 7.2, DNAStar, Madison, WI). When aligned, the 16 HA subtypes formed subclades with strong boot-trap support as previously reported (Air, 1981; Fouchier et al., 2005). To limit the number of primer sets needed for molecular subtyping, four pairs of consensus primers were designed from conserved regions of these subclades to amplify a 120 bp – 130 bp product for all 16 different HA subtypes (Table 1). Two pairs of the primers designed together amplify 10 of the 16 HA subtypes.
Table 1.
HA molecular subtyping RT-PCR primers designed for this study:
| Primer Name | Forward Primer | Reverse Primer | HA region | HA subtypes amplified |
|---|---|---|---|---|
| Primer Set 1 | 5′-TTC TAY AGR ARY ATS ARH TGG YTG AC-3′ | 5′-GGY GGR TGR TKD ATB CCC CA-3′ | HA11 | H3, H4, H8, H9, H12, H14 |
| Primer Set 2 | 5′-ATH GAV AAR ATG AAY ACH CAR TT-3′ | 5′-CNG CAT TRT AWG TCC ANA YDT C-3′ | HA22 | H1, H2, H5, H6, H12 |
| Primer Set 3 | 5′-TGG TTT AGC TTC GGG GMR TCA-3′ | 5′-ATA CAR ATD GTG CAY CGC A-3′ | HA23 | H7, H10, H15 |
| Primer Set 4 | 5′-GAG GNY TDT TYG GDG C-3′ | 5′-GAB KYY YTR TCT GCW GC-3′ | HA24 | H9, H11, H13, H16 |
Aligned to bases 516–634 of A/mallard/ALB/394/1988 (H3N3), GenBank accession No. CY005942
Aligned to bases 1230–1353 of A/mallard/ALB/267/1996 (H1N1), GenBank accession No. CY004504
Aligned to bases of 1600–1700 of A/mallard/Alberta/34/2001(H7N1), GenBank accession No. CY005983
Aligned to bases of 1057–1177 of A/mallard/ALB/124/1991(H11N2), GenBank accession No. CY006003
Extensive alignments with published avian influenza virus HA sequences suggested that these primer sets should amplify most known sequences. For example, an alignment of the 277 full-length H7 sequences in GenBank (including 263 low pathogenicity and highly pathogenic avian H7 sequences, 12 equine H7, and 2 human H7 sequences) showed that 277 out of 277 match completely the forward primer of set 3 (Table 1), and 228 out of 277 matched the reverse primer. The remaining 49 had a one base mismatch in the 5′ end of the primer, and thus it was expected this primer set would amplify all published H7 strains. Similarly, of the 234 full-length H9 sequences in GenBank, 215 out of 234 matched completely the forward primer of set 1. The remaining 19 out of 234 had a one base mismatch at the 5′ end of the primer and were also expected to be amplifiable with this primer set. The reverse primer matched completely in all 234 sequences. Lastly, an alignment of 359 full-length H5 sequences, representing isolates from years 1959–2006, and including both low pathogenicity and highly pathogenic H5 sequences from North America, Africa, Europe, and Asia, showed 328 out of 359 matching the forward primer of set 2 completely, while 31 out of 359 showed a one base mismatch. Only 6 of 359 had a one base mismatch in the reverse primer. Thus, all published H5 sequences were also expected to be amplifiable with primer set 2.
Reverse transcription and RT-PCR
RNA was purified from the swab samples with the MagMAX-96 viral isolation kit (Ambion, Foster City, CA) according to the manufacturer’s instructions and first strand cDNA was produced with 4 μl RNA lysate in a 20 μl reaction containing 200 U M-MLV reverse transcriptase (Invitrogen, Carlsbad, CA) following the manufacturer’s instructions.
RT-PCR was performed using 1 μl of the cDNA and one of the four pairs of primers was used individually for each reaction. Reactions were carried out in a final volume of 50 μl containing 1x PCR Gold buffer, 2.5 mM MgCl2, 0.2mM dNTP mix, 4U AmpliTaq Gold™ polymerase (Applied Biosystems, Foster City, CA) and each oligonucleotide primer at a concentration of 0.6 μM. Thermocycling was performed in a DNAEngine apparatus (Bio-RAD, Hercules, CA) with the following cycling conditions: 10 min at 95° C followed by 40 cycles of 94° C for 1 min, 50° C or 52° C for 1 min, and 70 °C for 1 min. The annealing temperature varied between the primer pairs: 52° C for primer set 1 and primer set 3 and 50° C for primer set 2 and primer set 4. All PCR reactions were performed with positive and negative controls. Influenza A virus RNA of known HA subtype (H1–H13) served as positive controls (H14–H16 controls were not available) in each case. After each reaction, a 5 μl portion of the PCR product was electrophoresed on a 2% TBE agarose gel stained with ethidium bromide, and visualized on a UV transilluminator.
Positive control RNA was extracted from avian influenza viruses sequenced as part of the NIAID genome sequencing project (http://www.niaid.nih.gov/dmid/genomes/mscs/influenza.htm) using strains from The Ohio State University repository (http://www.ncbi.nlm.nih.gov/genomes/FLU/Database/shipment.cgi) and consisted of representatives of HA subtypes H1–H8, H10–H12. Strains used were: A/mallard/Ohio/56/1999 (H1N1), A/mallard/Ohio/37/1986 (H2N1), A/mallard/Ohio/48/1986 (H3N2), A/mallard/Ohio/324/1988 (H4N6), A/mallard/Maryland/789/2002 (H5N2), A/black duck/Ohio/307/1987 (H6N2), A/mallard/Ohio/322/1998 (H7N3), A/mallard/Alaska/708/2005 (H8N4), A/long-tailed duck/Maryland/295/2005 (H10N8), A/black duck/Ohio/194/1986 (H11N1), A/mallard/Ohio/409/1988 (H12N5). A/chicken/Hong Kong/G9/1997 (H9N2), and A/gull/Maryland/704/1977(H13N6) were kindly provided by Dr. Kanta Subbarao (NIH). HA sequences from these strains are available in the Influenza Virus Resource page of the NCBI (http://www.nccbi.nlm.nih.gov/genomes/FLU/).
Cloning and Nucleotide sequencing
PCR products were either directly cloned into TOPO TA (Invitrogen, Carlsbad, California) or gel purified using an Ultrafree-DA column (Millipore, Billerica, MA) and then cloned into TOPO TA following the manufacturer’s instructions. Colonies were used as templates for PCR; PCR products were then subjected to standard DNA sequencing reactions. Sequences obtained were subjected to analysis using BLAST (http://www.ncbi.nlm.nih.gov/sutils/blast_table.cgi?taxid=11308&selectall) and via alignments of HA sequences downloaded from the Influenza Virus Resource page of the NCBI. Sequences were aligned using the Megalign program (Lasergene 7.2, DNAStar, Madison, WI). Phylogenetic trees were generated using this program and bootstrapped 500 times. Representative sequences obtained have been deposited in GenBank (accession numbers EU264565-EU264598).
Results and Discussion
Design of degenerate primer sets covering the 16 avian influenza A HA subtypes
Four degenerate primer sets were designed based on alignments of the 16 different HA subtype sequences, and theoretically these primers collectively should amplify all known subtypes (Table 1). To demonstrate the specificity of these primer sets for molecular HA subtyping, positive controls were tested. These PCR results matched the expected sequence alignments for all the HA subtypes (H1–H13) as predicted (Figure 1). Primer set 2, designed to amplify H1, H2, H5, and H6, also amplified H12, while primer set 4, designed to amplify H11, H13, and H16, also amplified H9 (see Figure 1). Positive controls representing subtypes H14, H15 and H16 were not available. Because these samples were obtained from North American ducks, we also did not expect to detect these subtypes (Krauss et al., 2004).
Figure 1.
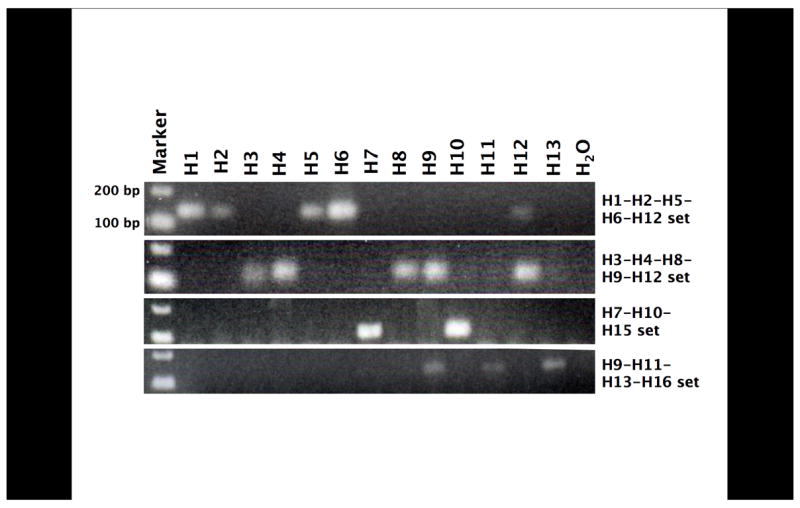
Positive control amplification with the 4 primer sets. cDNA from AIV of known subtype (H1–H13) were amplified with each of the 4 primer sets. PCR products were then electrophoresed on a 2% TBE agarose gel, stained with ethidium bromide, and photographed under ultraviolet transillumination.
Assay sensitivity
The assay reported here was designed to be a qualitative one, but the lower limit of detection was determined. Ten-fold serial dilutions with known quantities of influenza viral cDNA were performed. The assay primer sets have a lower limit of detection of 4–12 pg of input cDNA (data not shown). Thus, this assay is approximately 10–30-fold less sensitive than the matrix gene real-time RT-PCR (TaqMan) assay previously developed (Krafft et al., 2005) which was shown to have a detection limit of approximately 0.4 pg of input cDNA.
RT-PCR screening
Of 880 ethanol-fixed cloacal swabs analyzed previously for influenza A virus RNA, 225 were found to be positive (Runstadler et al., 2007). RNA from 222 of these positive samples was available for this molecular epidemiology study. Additionally, AIV was isolated from a subset of 42 birds for which paired cloacal swabs stored in viral transport medium were obtained. Of these, 38 were subtyped as described (Runstadler et al., 2007), and H3, H4, H8, and H12 subtypes were identified. Based on these results, the fixed swab samples were screened initially with primer set 1 and 193 of 222 (86.9%) of samples tested positive. RT-PCR products from 155 of 193 positive samples were cloned and multiple clones per insert were sequenced. Sequencing results revealed 7 H3, 2 H4, 21 H8, and 100 H12 subtypes, and 25 mixed samples (2 H3/H8, 5 H3/H12, 16 H8/H12, and 2 H3/H8/H12). Subsequently, 80 samples (representing 30 negative samples and 50 positive samples using primer set 1) were tested with primer set 2 and 50 RT-PCR positives were obtained. Of these, 43 were sequenced and revealed 15 H1, 1 H5, 3 H6 and 16 H12 in addition to 8 mixed samples (1 H1/H6, 5 H1/H12, 1 H1/H5/H6, and 1 H1/H5/H12). Twenty-three samples testing negative with both primer sets 1 and 2 were tested with primer sets 3 and 4, yet no additional positives were identified. A summary of the data is shown in Figure 2.
Figure 2.
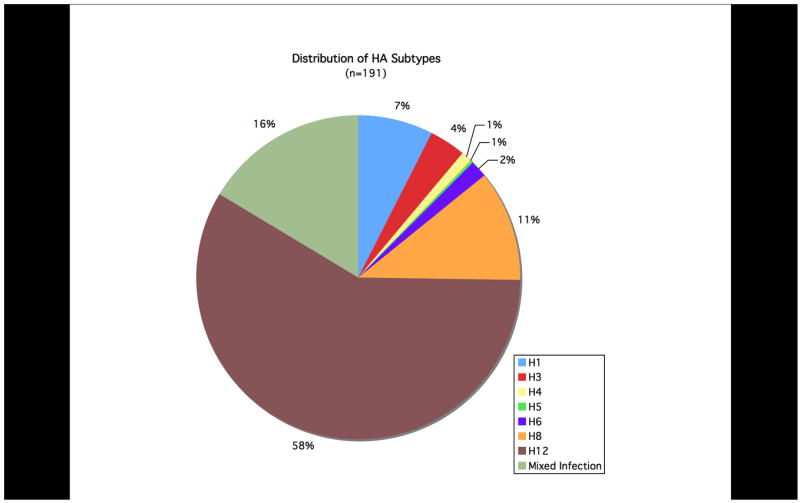
The percentage distribution of HA subtypes obtained from 191 ethanol-fixed cloacal swabs.
Of the 222 ethanol-fixed cloacal swab samples available for this study that were previously shown to be positive for influenza A virus RNA using a real-time TaqMan matrix gene assay (Runstadler et al., 2007), HA sequences were obtained from 191. Twenty-three samples tested negative with all four HA primer sets. The difference between samples identified as influenza virus RNA-positive using the matrix TaqMan assay but negative for the HA subtyping primer sets described here is likely due to assay sensitivity as described above.
Sequence analysis
BLAST analyses of the sequences revealed seven different co-circulating HA subtypes: H1, H3, H4, H5, H6, H8, and H12. As shown in Figure 2, H8, and H12 were most frequently detected, consistent with results from previous viral isolation attempts during this surveillance study. To further confirm the specificity of the subtypes identified through these primer sets, we designed additional primer sets targeting different regions of the HA gene segment and performed RT-PCR with these primers on 10 samples as a further control. Products were cloned and sequenced and in all cases matched the subtypes previously identified (data not shown). Sequences obtained from the samples are shown aligned to representative sequences downloaded from GenBank (Figures 3A and 3B). Representative full-length HA open reading frames of H1–H16 were aligned and analyzed phylogenetically. When the inter-primer sequences obtained from this study (approximately 75 bases) were added to the alignments, these small fragments clustered with the matching HA subtype with high bootstrap support (Figure 4).
Figure 3.


Sequence alignments. (A) Sequence alignments with the H1, H2, H5, H6, and H12 primer set. Representative HA genes from GenBank were aligned to the interprimer sequences obtained in this study. Sequences overlapping the degenerate primers are shown for the control sequences. (B) A similar alignment is shown for the H3, H4, H8, H9, H12, H14 primer set. Gaps were placed in the interprimer sequences to maximize the alignment.
Figure 4.


Phylogenetic trees of HA subtypes open reading frames plus sequence fragments from this study. (A) H1–H16 genes were aligned with the H1, H5, and H6 HA2 domain sequence fragments from this study using the Megalign program (Lasergene 7.2, DNAStar, Madison, WI) and bootstrapped 500 times. Bootstrap values for each of the HA subtypes are shown. The HA gene from the influenza B virus strain B/Lee/40 was used as an outgroup. (B) H1–H16 genes were aligned with the H3, H4, H8, and H12 HA1 domain sequence fragments from this study using the Megalign program (Lasergene 7.2, DNAStar, Madison, WI) and bootstrapped 500 times. Bootstrap values for each HA subtype are shown. The HA gene from the influenza B virus strain B/Lee/40 was used as an outgroup.
H5 subtype sequences
Three H5 sequences were obtained, and these sequences all closely matched H5 sequences from birds in North America (Figure 5) and were readily distinguishable from the highly pathogenic H5N1 viruses circulating in Eurasia. One sample was only positive for H5, but two H5 sequences were identified in samples showing mixed infections (in one case H1, H5, and H12 were recovered, and in another case H1, H3, H5, H6, and H12). Primer set 2 could amplify an Asian highly pathogenic H5N1 HA sequence used as a control (data not shown). The 2005 Alaska H5 sequences from this study share between 90–100% nucleotide identity with other North American lineage H5 sequences, but only approximately 75% identity with recent Eurasian highly pathogenic H5N1 sequences over this gene fragment. When full-length H5 HA open reading frames are aligned for comparison, North American H5 genes share between 90–100% sequence identity and 75–79% sequence identity with Eurasian highly pathogenic H5N1 strains, further validating the applicability of sequence analysis of the small inter-primer fragments obtained with these PCR sets.
Figure 5.

Sequence alignment of H5 sequences from this study with H5 sequences from North America and representative sequences of recent Eurasian-lineage highly pathogenic H5N1 H5 sequences (all downloaded from the Influenza Virus Resource).
Mixed infections
One of the interesting findings in this study of primary cloacal swab samples is that 2 or even up to 5 different HA subtypes were identified together in 16% of the samples, indicating a high rate of co-infection. Because the primer sets could amplify several different HA subtypes, multiple colonies (usually 8–10 colonies) were picked from the same plate for sequencing, and the large number of mixed infections highlight the utility of this molecular surveillance approach. Most notably, one ethanol-fixed swab sample, tested with both primer sets, was positive for H1, H3, H5, H6, plus H12. This bird also had a paired swab in viral transport media for culture and only an H3N8 virus was isolated and serotyped (A/pintail/Alaska/779/2005 (H3N8)). Four other samples with more than one HA subtype as determined by RT-PCR and sequencing also had matching cultured and serotyped viruses representing one of the determined subtypes in each case. Because all samples were not screened with all primer sets, the true number of mixed infections is likely to be even higher than the 16% detected here. A higher prevalence of mixed infections than previously thought may be an important contributing factor in the persistence of antigenic and genetic diversity among AIVs in wild bird populations.
Conclusions
We have previously shown that cloacal swabs fixed in ethanol could be analyzed by RT-PCR for the influenza A virus matrix gene (Runstadler et al., 2007) but that amplification template sizes needed to be under 200 bp (Evers, Slemons, and Taubenberger, 2007). Since previously published methods for molecular HA subtyping use either full-length amplicons (Bragstad et al., 2005; Hoffmann et al., 2001) or 640 bp HA2 domain templates (Phipps, Essen, and Brown, 2004), we reasoned that primer sets that would allow subtyping within the amplification constraints of the fixed swabs would be a useful adjunct to ongoing field surveillance studies for AIV molecular epidemiology. The method presented here allows molecular HA subtyping with a very small template. Because of the enormous sequence diversity between different HA subtypes, even the small inter-primer sequences analyzed here cluster with their cognate full-length gene sequences with high bootstrap values, supporting the robustness of this methodology. Given the high degree of variability of influenza A virus gene sequences, it is likely that these HA subtyping primer sets will not work with all sequences, especially if base-pairing mismatches occur at the 3′ ends of the primers, but the data reported here demonstrate that this method is robust not only using high quality viral RNA from the control samples but for RNA isolated from ethanol-fixed cloacal swabs in a remote field setting.
In addition to the possible expansion of field isolates that can be sampled with the fixed swab technique, molecular subtyping allows a direct examination of the complex co-circulation of different AIV HA subtypes present in a wild bird without the possible culture-induced bias of particular subtypes or strains out-growing others in a mixed infection in culture. We anticipate that this work will assist influenza A virus surveillance efforts to become more widespread, rapid, and inexpensive.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH and the NIAID.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Air GM. Sequence relationships among the hemagglutinin genes of 12 subtypes of influenza A virus. Proc Natl Acad Sci U S A. 1981;78(12):7639–43. doi: 10.1073/pnas.78.12.7639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DJ. An overview of the epidemiology of avian influenza. Vaccine. 2006 doi: 10.1016/j.vaccine.2006.10.051. [DOI] [PubMed] [Google Scholar]
- Bragstad K, Jorgensen PH, Handberg K, Hammer AS, Kabell S, Fomsgaard A. First introduction of highly pathogenic H5N1 avian influenza A viruses in wild and domestic birds in Denmark, Northern Europe. Virol J. 2007;4:43. doi: 10.1186/1743-422X-4-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragstad K, Jorgensen PH, Handberg KJ, Mellergaard S, Corbet S, Fomsgaard A. New avian influenza A virus subtype combination H5N7 identified in Danish mallard ducks. Virus Res. 2005;109(2):181–90. doi: 10.1016/j.virusres.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Evers DL, Slemons RD, Taubenberger JK. Effect of preservative on recoverable RT-PCR amplicon length from influenza A virus in bird feces. Avian Dis. 2007;51(4):965–68. doi: 10.1637/7880-012207-RESNOTER1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouchier RA, Munster V, Wallensten A, Bestebroer TM, Herfst S, Smith D, Rimmelzwaan GF, Olsen B, Osterhaus AD. Characterization of a novel influenza A virus hemagglutinin subtype (H16) obtained from black-headed gulls. J Virol. 2005;79(5):2814–22. doi: 10.1128/JVI.79.5.2814-2822.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson BA, Stallknecht DE, Swayne DE, Lewis LA, Senne DA. Avian influenza viruses in Minnesota ducks during 1998–2000. Avian Dis. 2003;47(3 Suppl):867–71. doi: 10.1637/0005-2086-47.s3.867. [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol. 2001;146(12):2275–89. doi: 10.1007/s007050170002. [DOI] [PubMed] [Google Scholar]
- Kawaoka Y, Krauss S, Webster RG. Avian-to-human transmission of the PB1 gene of influenza A viruses in the 1957 and 1968 pandemics. J Virol. 1989;63(11):4603–8. doi: 10.1128/jvi.63.11.4603-4608.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krafft AE, Russell KL, Hawksworth AW, McCall S, Irvine M, Daum LT, Connoly JL, Reid AH, Gaydos JC, Taubenberger JK. Evaluation of PCR testing of ethanol-fixed nasal swab specimens as an augmented surveillance strategy for influenza virus and adenovirus identification. J Clin Microbiol. 2005;43(4):1768–75. doi: 10.1128/JCM.43.4.1768-1775.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss S, Walker D, Pryor SP, Niles L, Chenghong L, Hinshaw VS, Webster RG. Influenza A viruses of migrating wild aquatic birds in North America. Vector Borne Zoonotic Dis. 2004;4(3):177–89. doi: 10.1089/vbz.2004.4.177. [DOI] [PubMed] [Google Scholar]
- Lee CW, Senne DA, Suarez DL. Development and application of reference antisera against 15 hemagglutinin subtypes of influenza virus by DNA vaccination of chickens. Clin Vaccine Immunol. 2006;13(3):395–402. doi: 10.1128/CVI.13.3.395-402.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munster VJ, Baas C, Lexmond P, Waldenstrom J, Wallensten A, Fransson T, Rimmelzwaan GF, Beyer WE, Schutten M, Olsen B, Osterhaus AD, Fouchier RA. Spatial, temporal, and species variation in prevalence of influenza A viruses in wild migratory birds. PLoS Pathog. 2007;3(5):e61. doi: 10.1371/journal.ppat.0030061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong WT, Omar AR, Ideris A, Hassan SS. Development of a multiplex real-time PCR assay using SYBR Green 1 chemistry for simultaneous detection and subtyping of H9N2 influenza virus type A. J Virol Methods. 2007;144(1–2):57–64. doi: 10.1016/j.jviromet.2007.03.019. [DOI] [PubMed] [Google Scholar]
- Payungporn S, Chutinimitkul S, Chaisingh A, Damrongwantanapokin S, Buranathai C, Amonsin A, Theamboonlers A, Poovorawan Y. Single step multiplex real-time RT-PCR for H5N1 influenza A virus detection. J Virol Methods. 2006;131(2):143–7. doi: 10.1016/j.jviromet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Phipps LP, Essen SC, Brown IH. Genetic subtyping of influenza A viruses using RT-PCR with a single set of primers based on conserved sequences within the HA2 coding region. J Virol Methods. 2004;122(1):119–22. doi: 10.1016/j.jviromet.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Runstadler JA, Happ GM, Slemons RD, Sheng ZM, Gundlach N, Petrula M, Senne D, Nolting J, Evers DL, Modrell A, Huson H, Hills S, Rothe T, Marr T, Taubenberger JK. Using RRT-PCR analysis and virus isolation to determine the prevalence of avian influenza virus infections in ducks at Minto Flats State Game Refuge, Alaska, during August 2005. Arch Virol. 2007;152(10):1901–10. doi: 10.1007/s00705-007-0994-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholtissek C, Rohde W, Von Hoyningen V, Rott R. On the origin of the human influenza virus subtypes H2N2 and H3N2. Virology. 1978;87(1):13–20. doi: 10.1016/0042-6822(78)90153-8. [DOI] [PubMed] [Google Scholar]
- Slemons RD, Hansen WR, Converse KA, Senne DA. Type A influenza virus surveillance in free-flying, nonmigratory ducks residing on the eastern shore of Maryland. Avian Dis. 2003;47(3 Suppl):1107–10. doi: 10.1637/0005-2086-47.s3.1107. [DOI] [PubMed] [Google Scholar]
- Slemons RD, Johnson DC, Osborn JS, Hayes F. Type-A influenza viruses isolated from wild free-flying ducks in California. Avian Dis. 1974;18(1):119–24. [PubMed] [Google Scholar]
- Spackman E, Senne DA, Myers TJ, Bulaga LL, Garber LP, Perdue ML, Lohman K, Daum LT, Suarez DL. Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J Clin Microbiol. 2002;40(9):3256–60. doi: 10.1128/JCM.40.9.3256-3260.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spackman E, Stallknecht DE, Slemons RD, Winker K, Suarez DL, Scott M, Swayne DE. Phylogenetic analyses of type A influenza genes in natural reservoir species in North America reveals genetic variation. Virus Res. 2005;114(1–2):89–100. doi: 10.1016/j.virusres.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Stallknecht DE, Shane SM. Host range of avian influenza virus in free-living birds. Vet Res Commun. 1988;12(2–3):125–41. doi: 10.1007/BF00362792. [DOI] [PubMed] [Google Scholar]
- Stone B, Burrows J, Schepetiuk S, Higgins G, Hampson A, Shaw R, Kok T. Rapid detection and simultaneous subtype differentiation of influenza A viruses by real time PCR. J Virol Methods. 2004;117(2):103–12. doi: 10.1016/j.jviromet.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Taubenberger JK, Morens DM, Fauci AS. The next influenza pandemic: can it be predicted? JAMA. 2007;297:2025–2027. doi: 10.1001/jama.297.18.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, Reid AH, Lourens RM, Wang R, Jin G, Fanning TG. Characterization of the 1918 influenza virus polymerase genes. Nature. 2005;437(7060):889–93. doi: 10.1038/nature04230. [DOI] [PubMed] [Google Scholar]
- Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. Evolution and ecology of influenza A viruses. Microbiol Rev. 1992;56(1):152–79. doi: 10.1128/mr.56.1.152-179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. World Health Organization; Geneva: 2007. Cumulative Number of Confirmed Human Cases of Avian Influenza A/(H5N1) Reported to WHO, Vol. 2007. [Google Scholar]