

Summary
Immune homeostasis is essential for the normal functioning of the immune system and its breakdown leads to fatal inflammatory diseases. We report here the identification of a novel member of the tumor necrosis factor-α-induced protein-8 (TNFAIP8) family, designated TIPE2 that is required for maintaining immune homeostasis. TIPE2 is preferentially expressed in lymphoid tissues and its deletion in mice leads to multi-organ inflammation, splenomegaly and premature death. TIPE2-deficient animals are hypersensitive to septic shock, and TIPE2-deficient cells are hyper-responsive to Toll-like receptor (TLR) and T cell receptor (TCR) activation. Importantly, TIPE2 binds to caspase-8, and inhibits activating protein-1 and nuclear factor-κB activation while promoting Fas-induced apoptosis. Inhibiting caspase-8 significantly blocks the hyper-responsiveness of TIPE2-deficient cells. These results establish that TIPE2 is an essential negative regulator of TLR and TCR function, and its selective expression in the immune system prevents hyper-responsiveness and maintains immune homeostasis.
Keywords: immune homeostasis, inflammation, Toll-like receptors, apoptosis, caspase
Introduction
The immune system is frequently bombarded by large quantities of foreign antigens. Upon engaging these antigens through specific receptors, the immune system mounts specific responses characterized by immune cell activation, proliferation and inflammatory gene expression, which in turn lead to inflammation. Upon removal of the antigen, the immune system returns to its pre-activation state, ready to respond to new antigens. The strength and the duration of the immune responses must be tightly regulated because uncontrolled inflammation can cause severe tissue injury and death of the organism. Immune homeostasis is the inherent property of the immune system to maintain a constant number of immune cells and to prevent deleterious inflammatory responses despite frequent stimulations by antigens. It ensures that any antigen-precipitated change of the immune system is kept to the minimum so that the immune responses eliminate the antigen in question but do not lead to fatal inflammatory diseases (Van Parijs and Abbas, 1998). The molecular mechanisms through which immune homeostasis is maintained are not fully understood, but recent studies indicate that at least two classes of molecules are required. The first class includes molecules that limit the strength of immune cell activation and expansion. These include inhibitory cytokines [such as transforming growth factor (TGF)-β and interleukin (IL)-10], negative regulators of the antigen receptor and Toll-like receptor (TLR) signaling (such as CTLA-4, inhibitor-of-κB, Socs1 and itch) and repressive transcription factors (such as Foxp3) (Van Parijs and Abbas, 1998; Wahl et al., 2006). The second class controls cell death, which includes Fas, Bim, Bax, caspases-8 and 10 (Nagata and Suda, 1995; Van Parijs and Abbas, 1998). When these molecules are depleted from the immune system, immune homeostasis fails and severe inflammatory diseases ensue which in turn lead to premature death of the organism (Van Parijs and Abbas, 1998). Because a large number of genes in the mammalian genomes have not yet been characterized, the number and identity of additional genes required for maintaining immune homeostasis are unknown.
Tumor necrosis factor (TNF)-α-induced protein 8 (TNFAIP8), also known as SCC-S2, GG2-1 and MDC-3.13, is a newly identified apoptosis regulator (Kumar et al., 2004; Kumar et al., 2000; Patel et al., 1997; Yoshinaga-Hirabayashi and Yoneda, 2001; Zhang et al., 2006). Like several other regulators/mediators of apoptosis, TNFAIP8 contains a death effector domain (DED) and is able to inhibit caspase-mediated apoptosis (Kumar et al., 2000). Knocking down TNFAIP8 expression in tumor cells reduces their tumorigenicity suggesting that it may play a role in oncogenesis (Zhang et al., 2006). However, the physiological role of TNFAIP8 is not clear. In an attempt to identify novel genes that regulate inflammation and immune homeostasis, we analyzed the transcriptome of murine spinal cord before and after the development of autoimmune encephalomyelitis, using a high throughput gene microarray technology. We identified more than a hundred novel genes that are highly expressed in inflamed but not normal nervous tissue (Carmody et al., 2002). One of these genes shares a high degree of sequence homology with TNFAIP8, which we call TIPE2 (tumor necrosis factor-α-induced protein 8-like 2). Using knockout, knockdown and gain-of-function approaches, we established that TIPE2 is a novel negative regulator of immune cell function, required for preventing hyper-responsiveness and maintaining immune homeostasis.
Results
Identification of a novel member of the TNFAIP8 family from inflamed spinal cord
To isolate new genes involved in autoimmune inflammation, we used Affymetrix gene chips to examine gene expression profiles of murine spinal cords before and after the development of experimental autoimmune encephalomyelitis (EAE) (Carmody et al., 2002). This led to the identification of 141 novel genes that were highly expressed in the inflamed spinal cord but totally absent in normal spinal cord (Carmody et al., 2002). One of these genes was represented in the gene bank as the expressed sequence tag (EST) AI847688. By aligning this EST against the mouse EST database in the gene bank using the BLAST sequence homology search program, we identified a putative full-length cDNA, designated TIPE2 (TNFAIP8L2), with a predicted open reading frame encoding 184 amino acids (Figure S1A). By aligning this sequence against the human EST database in the gene bank using the same BLAST program, we identified a putative TIPE2 human homologue that shares 94% amino acid sequence identity with murine TIPE2 (Figure S1A). Human TIPE2 shares approximately 53% identity and 78% similarity with TNFAIP8 (Kumar et al., 2000). Additionally, TIPE2 contains a putative DED-like domain that shows significant identity/similarity to other known DED sequences (Figure S1B). The identity/similarity shared between DED of murine TIPE2 and those of the following proteins are as follows: TNFAIP8, 50%/73%; murine cFLIP DED I, 19%/32%; murine cFLIP DED II, 13%/33%; murine caspase-8 DED I, 19%/40%; murine caspase-8 DED II, 20%/38%. The TIPE2 DED domain resides in its NH2-terminal region (Figure S1). Like other DED-containing proteins, TIPE2 also possesses six putative conserved α helices as determined by NPS Network Protein Sequence Analysis (Combet et al., 2000). Besides TIPE2, two additional members of the TNFAIP8 family may exist, which share high degrees of sequence homology with TIPE2 and are designated in the gene bank as TNFAIP8L1 (TIPE1) and TNFAIP8L3 (TIPE3). Thus, the TNFAIP8 family may consist of at least four members.
The chromosome location of TIPE2 was then determined by aligning the murine and human TIPE2 sequences with murine and human genome databases, respectively. A single locus was identified for murine TIPE2 on chromosome III (3f1–3f3) and for human TIPE2 on chromosome I (1q21.2–1q21.3).
Preferential expression of TIPE2 in lymphoid and inflamed tissues
To determine the expression pattern of TIPE2, total RNA was extracted from tissues of perfused normal and EAE mice, fractionated by electrophoresis and transferred to a Hybond-N nylon membrane. The RNA was then hybridized with the full-length TIPE2 cDNA probe. A ~1.1 kb transcript was detected in the thymus, spleen, lymph node and small intestine, but not in the liver, heart, muscle, testis, spinal cord or brain of normal mice. By contrast, high levels of TIPE2 mRNA were detected in the spinal cord of mice with EAE (Fig. 1A). Furthermore, a weak TIPE2 signal was also detected in the lung, skin and colon, which all contain lymphoid tissues. Therefore, TIPE2 detected in the inflamed spinal cord is likely expressed by infiltrating cells of the immune system.
Figure 1. Preferential expression of TIPE2 in lymphoid tissues and inflamed spinal cord.
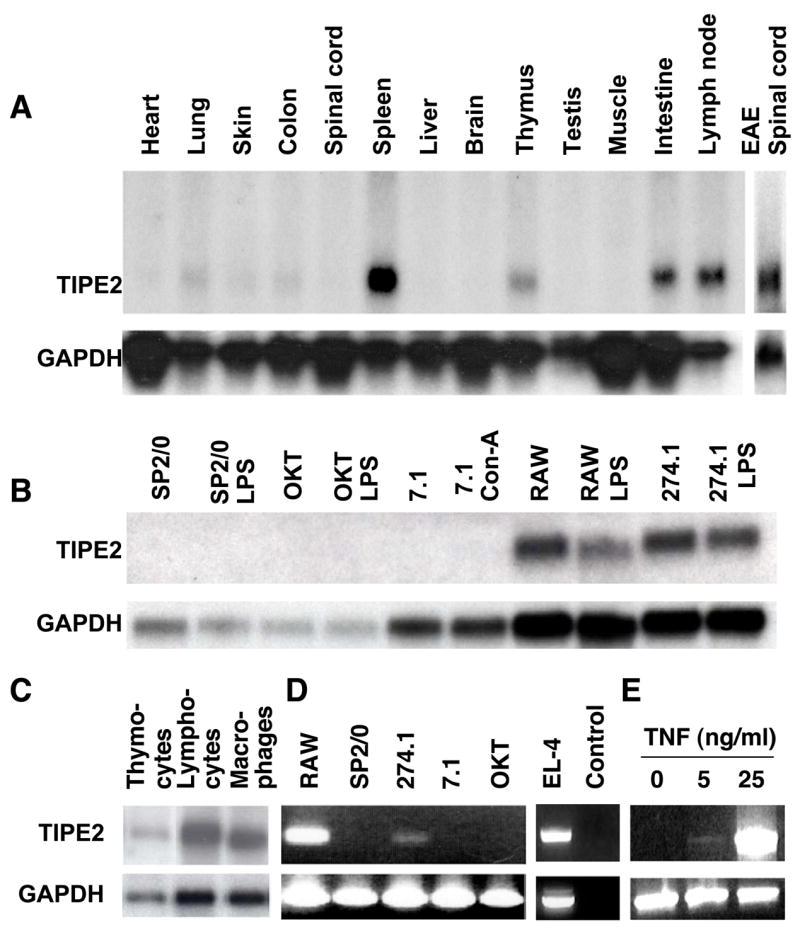
Northern blot (A–C) and RT-PCR analyses (D–E) of TIPE2 expression in selected tissues and cell preparations. A. RNAs were extracted from freshly harvested organs of either normal C57BL/6 mice (first 13 lanes) or mice with EAE (last lane). B. RNAs were extracted from murine cell lines that were pretreated with or without 2 μg/ml of concanavalin (Con)-A, or 2 μg/ml of lipopolysacchrides (LPS) for 24 hrs. OKT, OKT-3 B cells. C. RNAs were extracted from the following cell types of the C57BL/6 mice: lane 1, total thymocytes; lane 2, enriched splenic lymphocytes; lane 3, enriched splenic macrophages. D. RT-PCR analysis of total RNA extracted from murine cell lines using specific primers for TIPE2 and GAPDH. No template DNA was added to the control lane. E. NIH3T3 fibroblasts were cultured with 0–25 ng/ml of mouse TNF-α for 4 hrs. TIPE2 and GAPDH expression was determined by RT-PCR.
To determine which cell type expresses TIPE2, we performed Northern blot and/or PCR analysis of a panel of cell preparations (Fig. 1B-E). We found that macrophages, B and T lymphocytes of various developmental stages constitutively expressed TIPE2 (Fig. 1C and Fig. S2). One of the two T cell lines (EL-4) and two of the macrophage cell lines (RAW 264.7 and Wehi 274.1) expressed TIPE2. By contrast, neither NIH3T3 fibroblasts nor myeloma cell lines (SP2/0 and OKT3) constitutively expressed TIPE2. However, following stimulation with TNF-α, NIH3T3 fibroblasts expressed detectable levels of TIPE2 mRNA (Fig. 1E). Thus, TIPE2 is preferentially expressed by lymphoid and myeloid cells but may be induced in other cell types by TNF-α.
Spontaneous development of fatal inflammatory diseases in TIPE2-deficient mice
To elucidate the roles of TIPE2 in vivo, we generated TIPE2-deficient mice by deleting both exons of the TIPE2 gene through homologous recombination (Figure S3). The TIPE2 mRNA is completely absent in mice homozygous for the gene mutation (Figure S3C). Mice homozygous for the TIPE2 gene mutation developed normally and were born with the expected Mendelian ratio. However, starting from approximately 2 months of age, many TIPE2−/− mice became chronically ill, characterized by body weight loss, splenomegaly, leukocytosis, and multi-organ inflammation. By the end of the 11th month, ~50% of TIPE2−/− mice died of the disease (Fig. 2A). Histological examinations revealed severe mononuclear cell infiltration in multiple organs including lung (Fig. 2D), liver and intestine starting approximately three months after the birth. The splenic microarchitecture was also destroyed due to the enlargement of the white pulps. Serological testing showed high levels of inflammatory cytokines such as interleukin (IL)-1, IL-6, IL-12 and TNF-α as well as the inhibitory cytokine IL-10 in the blood of TIPE2−/− but not TIPE2+/+ mice (Fig. 2B). By contrast, the total amounts of immunoglobulin (Ig) of various isotypes remained largely unchanged in 4 month old TIPE2−/− mice with the exception of IgG2a and IgG2b which were reduced as compared to those of control littermates (Fig. S4). This indicates that class switching of certain Ig isotypes may be disrupted in aged TIPE2−/− mice, which could be secondary to the diseased conditions of these mice. Importantly, no increase in seral antibodies to single or double stranded DNA was detected in 4 month old TIPE2−/− mice. Splenomegaly and leukocytosis were caused by an increase in the numbers of both myeloid and lymphoid cells including CD11b+, B220+, and CD4+ cells (Fig. 3). A significantly greater number of splenocytes in TIPE2−/− mice expressed the early activation marker CD69 (Fig. 3D) as compared to wild type (WT) littermates, suggesting heightened cell activation. All mice were kept in a pathogen-free environment and recombination activating gene (RAG)-1-deficient mice that do not have lymphoid cells housed in the same environment did not develop spontaneous infectious diseases. We were unable to identify any infectious agents in sick TIPE2−/− mice by culture, histochemistry and routine serological testing.
Figure 2. Spontaneous development of fatal inflammatory diseases in TIPE2-deficient mice.
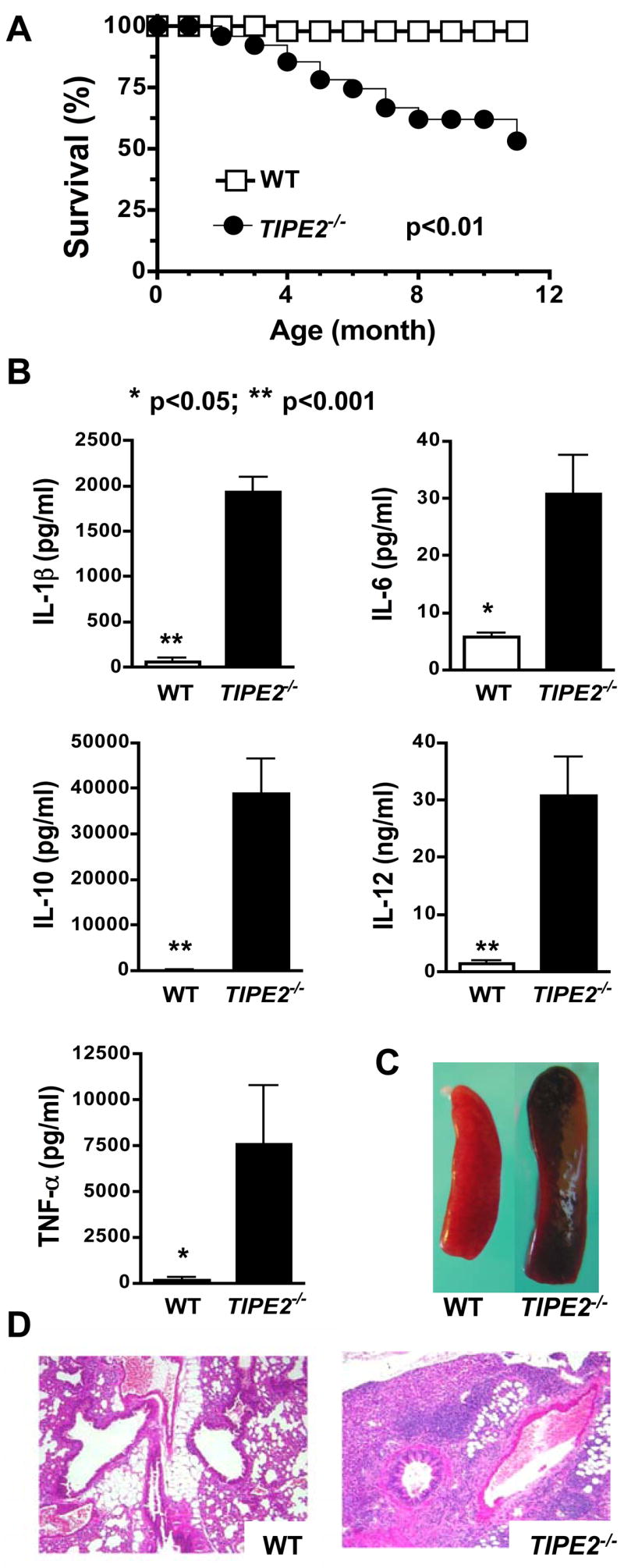
A. The survival curves of wild type (WT) (n=36) and TIPE2−/− littermates (n=36) over a period of 11 months. B. Increased seral cytokines in TIPE2-deficient mice. Sera were collected from 4-month-old wild type (n=6) and TIPE2−/− littermates (n=6) and tested for cytokines by ELISA. Data shown are means and SD of all mice, and are representative of three experiments. C. Increased spleen size in TIPE2-deficient mice. Spleens of a wild type and a TIPE2-deficient littermate at 4 months of age are shown. D. Severe lnterstitial lung inflammation in TIPE2-deficient mice. Lung sections of a wild type and a TIPE2-deficient littermate at 4 months of age are shown. Sections were stained with hematoxylin and eosin, and examined by microscopy with 200x magnifications.
Figure 3. Characteristics of TIPE2-deficient mice.
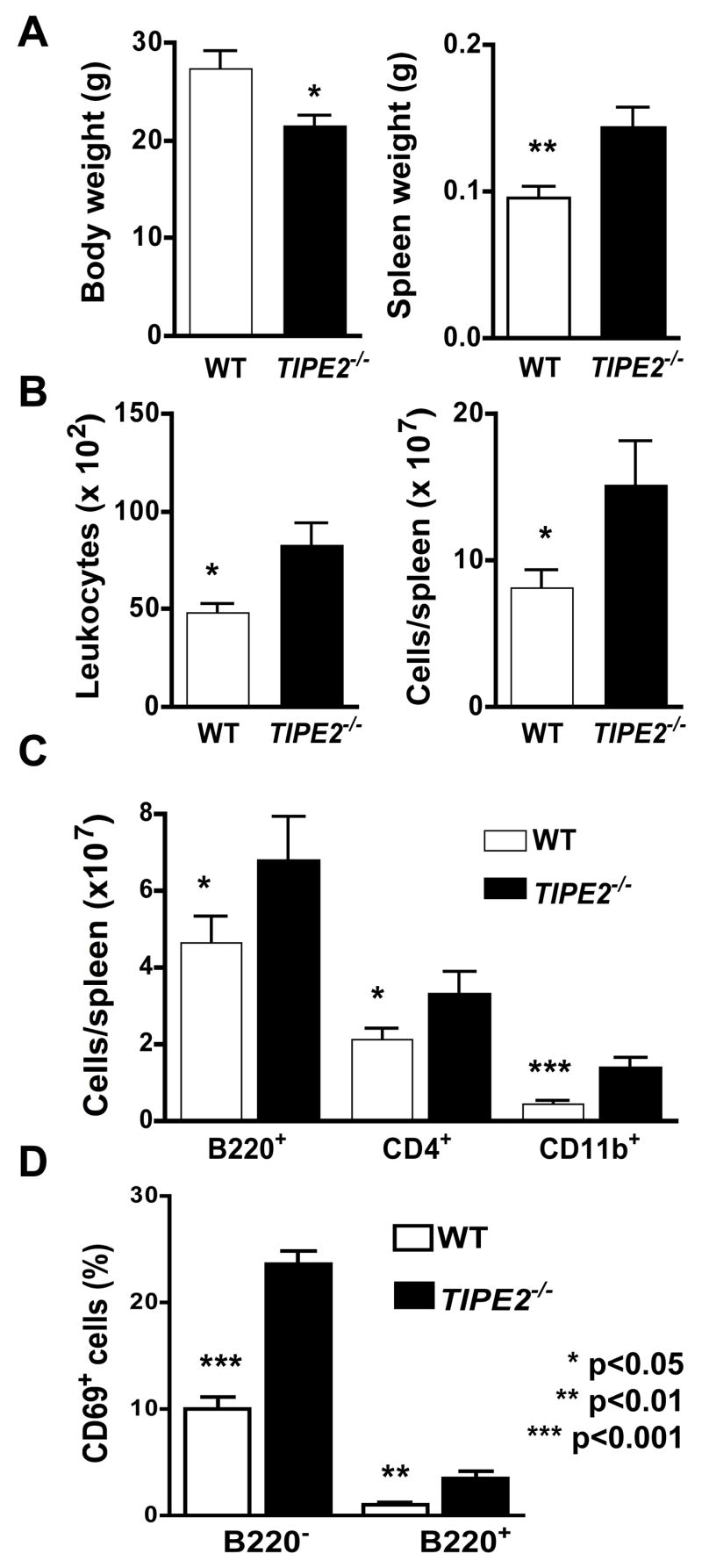
Wild type (n=11) and TIPE2-deficient littermates (n=11) were sacrificed at 4 months of age and tested for the following parameters. A. Body and spleen weights. B. Blood leukocyte counts and splenocyte counts. C. B220+, CD4+ and CD11b+ splenocyte counts by flow cytometry following staining cells with respective antibodies. D. Percentages of CD69+ splenocytes in B220+ and B220− sub-populations. Data shown are means and SD, and are representative of two experiments.
Negative regulation of T cell-mediated immunity by TIPE2
To determine whether TIPE2 gene mutation affects immunity, we studied humoral and cellular immune responses in TIPE2−/− mice using the following three experimental systems: 1) lymphocytic choriomeningitis virus (LCMV) infection (Fig. 4), 2) ovalbumin (OVA) immunization with complete Freund’s adjuvant (CFA) (Figs. S5), and 3) Listeria monocytogenes infection. We found that both CD4+ and CD8+ T cell immune responses were significantly augmented in TIPE2−/− mice as compared to their littermate controls. This was reflected in the number of LCMV-specific CD8+ T cells in the spleen (Fig. 4A and 4B), cytokines released by LCMV- or OVA-specific cells in the culture (Fig. 4C and Fig. S5), and cytokines in the sera of mice infected with Listeria(unpublished data). In contrast to the enhanced cellular immunity, humoral immunity in TIPE2−/− mice remained largely undisturbed. Immunization of young TIPE2−/− mice with OVA induced similar levels of OVA-specific antibodies as compared to their control littermates (unpublished data). Taken together, these results indicate that cellular but not humoral immunity is significantly disrupted in TIPE2−/− mice. As further discussed below, this dichotomy is likely due to the receptor-specific, but not cell-specific, functions of TIPE2.
Figure 4. Increased T cell immunity of TIPE2 knockout mice.

A-C. Increased T cell immune responses to LCMV infection. TIPE2−/− mice and their littermate controls (n=6), 5–6 weeks of age, were injected i.p. with 106 LCMV Armstrong, and sacrificed 8 days later. Splenocytes were collected, stained with anti-CD8 and LCMV-specific Db/gp33-tetramer, and examined by flow cytometry (A) (Ochsenbein et al., 1999). The total numbers of CD8+, Db/gp33-tetramer+ (tet+) cells per spleen were presented in 4B. Splenocytes were also cultured with LCMV-gp33–41 peptide (1μM) for 48 hours, and the concentrations of IFN-γ in the supernatants were determined by ELISA (C). Results are representative of two independent experiments. D. Increased reactivity of TIPE2 knockout T cells to TCR stimulation. CD4+ T cells were purified from spleens of 4–5-week-old wild type (n=5) or TIPE2-deficient littermates (n=5), and stimulated with indicated amounts of plate-bound anti-CD3 and 1 μg/ml of soluble anti-CD28 mAbs. Cytokine concentrations in the culture supernatants were determined by ELISA at 24 hr, and proliferation was measured by 3H-thymidine incorporation [presented as count per minute (CPM)] at 48 hr. Data shown are means and SD, and are representative of three experiments. No significant differences were detected between knockout and control groups in terms of the number and percentage of dead cells as determined by annexin V staining.
In vitro, purified TIPE2−/− T cells were hyper-reactive to TCR ligation and produced significantly higher levels of Th17 cytokines as compared to WT controls (Fig. 4D). The production of Th1 cytokine IFN-γ and Th2 cytokine IL-4 was also increased to various degrees in TIPE2−/− cultures, but no significant changes in T cell proliferation was apparent. In addition to cytokines, CD25, CD44 and CD69 expression was also higher in TIPE2 knockout T cells activated in Fig. 4D. Specifically, the percentages of T cells that expressed CD25, CD44 and CD69 at the end of the culture increased from 60±12, 50±11 and 64±11 in the wild type group to 87±1.4, 72±2 and 90±1.1 in the knockout group, respectively (p<0.02) (Fig. S6). Cells used in these experiments were from 4–5-week old mice that did not have detectable inflammatory diseases or increased numbers of CD69+ activated cells.
These results indicate that TIPE2 may negatively regulate T cell activation. To further test this theory, we utilized a retrovirus-mediated gene transfer system that allows stable TIPE2 gene transfer into hematopoietic cells. We found that the expression of the activation marker CD69 and cytokine IFN-γ was significantly reduced in cells over-expressing TIPE2 following stimulation with the specific antigen (Fig. S7). This was accompanied by a small but detectable reduction in the activation marker CD44 expression in these cells (Fig. S7). Furthermore, cell division was also reduced in cells over-expressing TIPE2, indicating that TIPE2 may regulate mitosis under certain conditions. Taken together, these results indicate that TIPE2 is able to inhibit TCR-mediated T cell activation.
Hypersensitivity of TIPE2 knockout and knockdown cells to Toll-like receptor stimulation
To determine the potential roles of TIPE2 in innate immune responses to TLR ligands, we studied TIPE2 knockout and knockdown cells in vitro. Upon stimulation with LPS, bone marrow-derived TIPE2−/− macrophages produced significantly more IL-6 and IL-12 than wild type cells, with relatively normal amounts of TNF-α (Fig. 5A). Similarly, knocking down TIPE2 expression in the RAW 264.7 macrophages significantly increased the levels of IL-6 protein (Fig. 5C) and mRNA (Fig. 5D). The effect of TIPE2 knockdown is not limited to LPS because responses to three additional TLR ligands were also significantly increased in the knockdown macrophages (Fig. 5E).
Figure 5. Increased reactivity of TIPE2 knockout and knockdown macrophages to Toll-like receptor stimulation.

A. Bone marrow-derived macrophages from wild type and TIPE2-deficient mice (n=5) were treated with or without LPS (100ng/ml) for 8 hours. IL-6, IL-12p40 and TNF-α concentrations were determined by ELISA. B. TIPE2 mRNA levels in wild type and TIPE2 knockdown (KD) RAW 264.7 macrophages as determined by RT-PCR. GAPDH was used as a loading control whereas H2O was used as the background control. C–D. Wild type and TIPE2 knockdown RAW 264.7 cells were treated with LPS (100ng/ml) for the indicated times. IL-6 protein (C) and mRNA (D) levels were determined by ELISA and real-time PCR, respectively. E. Wild type and TIPE2 knockdown RAW 264.7 cells were treated with LPS (100ng/ml), peptidoglycans (PGN) (10μg/ml), Poly(I:C) (50μg/ml), or CpG oligodeoxynucleotides (CPG) (1μM) for 8 hours. IL-6 concentrations in the supernatants were determined by ELISA. Data shown are representative of three experiments. The p value shown is for all TLR treated WT cultures as compared to their respective TIPE2 KD groups. No significant differences were detected between the two groups in terms of the number and percentage of apoptotic cells as determined by annexin V staining.
Furthermore, TIPE2−/− splenic B cells expressed significantly more IL-1β and TNF-α than wild type B cells upon stimulation with LPS or CpG (Fig. S8, A and B), indicating that the innate immune function of TIPE2−/− B cells is also augmented. By contrast, anti-IgM-induced TNF-α expression was not affected by TIPE2 deficiency (Fig. S8B). Proliferation, apoptosis and Ig secretion of B cells treated with anti-IgM were not significantly different between wild type and TIPE2−/− groups (unpublished data). Thus, TIPE2 may regulate innate but not adaptive B cell responses. This may explain why humoral immunity is not significantly disrupted in TIPE2−/− mice (Figs. S4). It is to be noted that TNF-α is significantly affected by TIPE2 deficiency in B cells but not in macrophages treated with TLR ligands. This indicates that TIPE2 may regulate TNF-α expression in a cell-specific manner. Differences between B cells and macrophages in their usage of transcription factors and signaling molecules are likely responsible for this cell-specific effect of TIPE2 on TNF-α.
Hypersensitivity of TIPE2 knockout mice to septic shock
If TIPE2 deficiency enhances TLR activation, will it subject mice to septic shock? To address this question, we studied LPS-induced sepsis in mice that were injected with a low dose LPS. We found that septic shock was dramatically accelerated and exacerbated in TIPE2−/− mice as compared to wild type controls. This was reflected by a sharp decline in the survival rate (0% vs. 91%), a significant increase in seral IL-1β, IL-6 and IL-12p40 (Fig. 6), and severe multi-organ necrosis in TIPE2−/− mice (data not shown). Because mice used in these experiments were only 5–6 week old, not suffering from the inflammatory syndrome described above, this data indicates that TIPE2 is directly responsible for preventing septic shock.
Figure 6. Hypersensitivity of TIPE2 knockout mice to septic shock.
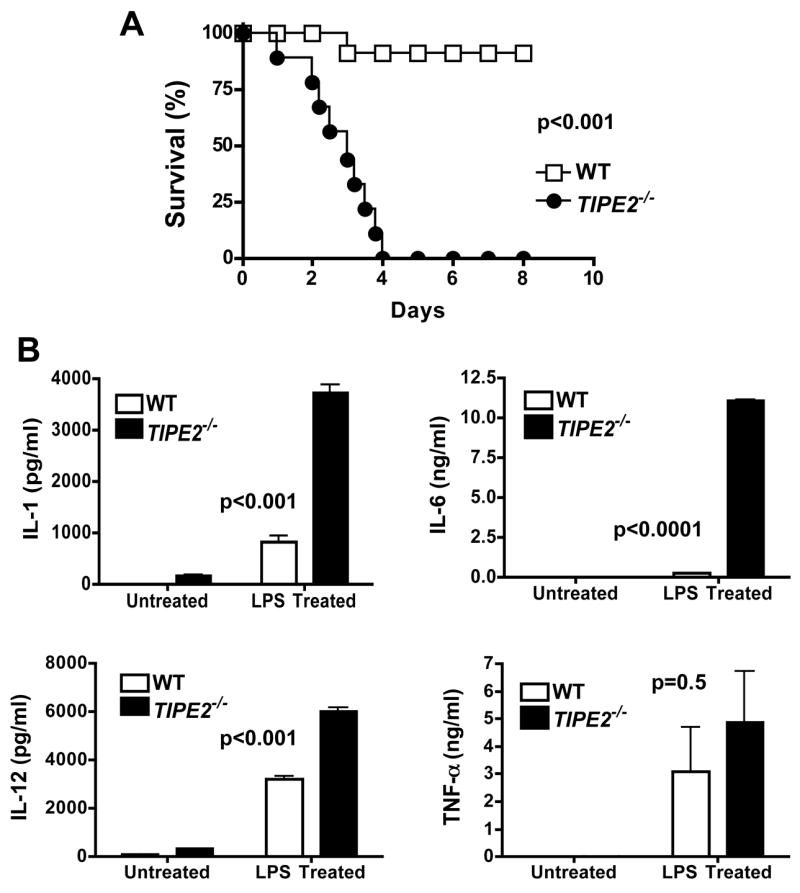
Wild type (n=11) and TIPE2-deficient littermates (n=10), 5–6 weeks of age, were injected intravenously with a low dose LPS (15 mg/kg). A. The survival curves. B. IL-1β, IL-6, IL-12p40 and TNF-α concentrations in the sera as determined by ELISA. For the LPS treated group, mice were injected with LPS as in panel A, and sera were collected 48 hrs later. For the untreated group, sera were collected from age and sex matched naïve mice (n=6). Data shown are means and SD, and are representative of two experiments.
Negative regulation of activating protein (AP)-1 and nuclear factor (NF)- κ B pathways by TIPE2
Most cytokines affected by TIPE2 deficiency or knockdown are targets of two major signaling pathways downstream of both TCR and TLR, i.e., the mitogen-activated protein (MAP) kinases and the NF-κB pathways. To determine whether TIPE2 regulates these two pathways, we performed biochemical analyses of key signaling molecules involved. We found that the c-Jun N-terminal kinase (JNK), p38 and the NF-κB pathway, but not the extracellular signal-regulated kinase (ERK) pathway, were targets of TIPE2 action. Specifically, TIPE2 knockdown amplified the JNK and p38 pathways at the level of AP-1 binding to DNA (Fig. S9A), c-Fos and c-Jun nuclear translocation (Fig. 7A), as well as JNK1/2 and p38 phosphorylation (Fig. 7A and 7B). Similarly, TIPE2 knockdown enhanced NF-κB nuclear translocation (Fig. S10) and inhibitor-of-κB (IκB) phosphorylation (Fig. 7A); NF-κB binding to DNA was also increased in resting TIPE2 knockdown cells suggesting that TIPE2 may regulate the basal levels of the NF-κB activity (Fig. S9B). By contrast, little or no difference in ERK activation was noted between wild type and TIPE2 knockdown cells (Fig. 7A). Consistent with these results, JNK and p38 activities were also significantly increased in TIPE2 knockout T cells as compared to wild type controls (Fig. S11A). In TIPE2 knockout macrophages, significant increases in IκBα degradation and phosphorylation were observed 15–60 mins after LPS stimulation, whereas increases in JNK and p38 phosphorylation were noted only at 5 and 120 mins after LPS stimulation (Fig. S11B). No significant differences in ERK phosphorylation between wild type and TIPE2 knockout macrophages were noted at any time points tested. Taken together, these data indicate that TIPE2 is a negative regulator of NF-κB, JNK and p38 pathways.
Figure 7. Mechanisms of TIPE2 action.

A-B. Increased AP-1 and NF-κB activation in TIPE2 knockdown cells. Wild type and TIPE2 knockdown RAW 264.7 cells were stimulated with LPS (100ng/ml) for the indicated times. c-Fos and c-Jun levels in the nuclear extracts were determined by Western blot, with histone H1 serving as a loading control (A, top two panels). Total cell lysates were also blotted with antibodies to total or phosphorylated (p) IκBα, p38, ERK (A, bottom three panels) and JNK1/2 (B). C. TIPE2 binds to endogenous caspase-8. Total cell lysates of wild type and TIPE2-knockdown RAW 264.7 cells were immunoprecipitated with an anti-TIPE2 rabbit polyclonal antibody or control rabbit Ig. The immunoprecipitates and cell lysates were then blotted with anti-caspase-8 (anti-casp-8), anti-TIPE2 or anti-FLIP antibodies. The level of TIPE2 in the crude lysate was too low to be detected by immunoblot (data not shown). D. Caspase-8 blockade diminishes the hyper-reactivity of TIPE2 knockdown cells. Wild type and TIPE2 knockdown RAW 264.7 cells were stimulated with LPS (100ng/ml) for 8 hrs with or without 1μg/ml of a caspase-8 inhibitor (Calbiochem). The level of intracellular IL-6 expression was determined by flow cytometry. Data shown are representative of two experiments. E–F. TIPE2 knockout T cells are resistant to activation-induced, but not cytokine withdrawal-induced, cell death. Splenocytes from wild type and TIPE2 knockout mice (n=5) were subjected to cytokine withdrawal-induced (E) or activation-induced (F) cell death as described in Procedures. The percentage of apoptotic cells was determined by flow cytometry following staining cells with annexin V, anti-CD4 and anti-CD8 antibodies (Song et al., 2000). Data shown are means and SD, and are representative of two experiments. G.TIPE2 knockdown in EL-4 T cells renders them resistant to FasL-induced apoptosis. TIPE2 mRNA levels in non-manipulated EL-4, control RNAi (wild type) and TIPE2 RNAi (knockdown) treated EL-4 cells were determined by RT-PCR (top panel). Wild type and TIPE2 knockdown EL-4 cells were either left untreated (control), or treated with 10 ng/ml FasL for 6 hrs (bottom panel). After staining with annexin V and propidium iodide (PI), cell death was analyzed by flow cytometry. Data shown are means and SD of the percentages of annexin V+ PI+ cells, and are representative of two experiments.
Caspase-8 interaction with endogenous TIPE2 and its role in TIPE2 function
Results discussed above indicate that the point of action of TIPE2 may lie upstream of MAPK and NF-κB pathways. Because TIPE2 has a DED-like domain and certain DED-containing proteins such as caspase-8 regulate NF-κB activation, we hypothesized that TIPE2 might interact with these proteins. To test this theory, the HA-or FLAG-tagged TIPE2, FLAG-tagged mutant procaspase-8 (the cysteine at position 360 was replaced by serine to abolish its apoptotic activity), FLAG-tagged Fas-associated death domain (FADD) were expressed in 293 cells following transfection with corresponding expression plasmids (Fig. S12). Co-immunoprecipitation analyses revealed that overexpressed TIPE2 specifically associated with over-expressed or endogenous caspase-8 but not FADD. To confirm this interaction for the endogenous TIPE2 protein, RAW 264.7 macrophage extracts were used to immunoprecipitate TIPE2-binding proteins with a specific anti-TIPE2 antibody. Upon blotting with anti-caspase-8 antibody, a strong caspase-8 signal was detected in the precipitates (Fig. 7C). This caspase-8 signal was almost completely absent in the TIPE2 knockdown cells, confirming the specificity of the assay. By contrast, FLIP was not detected in the immunoprecipitates. Robust interactions between endogenous TIPE2 and caspase-8 were observed in both resting and LPS-stimulated RAW cells, with little detectable differences. Therefore, TLR stimulation may not be required for TIPE2 binding to caspase-8. Importantly, both FADD and caspase-8 were normally recruited to the death-inducing signaling complex (DISC) (Fig. S13) in TIPE2 knockdown cells following Fas ligation. Caspase-8 cleavage was also normal in these cells. By contrast, TIPE2 was not recruited to the DISC (Fig. S13). Therefore, the target of TIPE2 action on the apoptotic pathway is likely downstream of caspase-8 or the DISC.
These results establish that TIPE2 is a novel caspase-8-binding protein, which may regulate caspase-8-dependent functions. In addition to inducing apoptosis, caspase-8 is also crucial for lymphocyte and myeloid cell activation, presumably through regulating the NF-κB signaling pathway (Bidere et al., 2006; Chaudhary et al., 1999; Lemmers et al., 2007; Su et al., 2005; Takahashi et al., 2006). Blocking caspase-8 activation with a specific caspase-8 inhibitor impedes the phosphorylation of IκBα, but not that of ERK, JNK or p38 (Fig. S14 and unpublished data). To determine whether caspase-8 played any role in TIPE2 functioning, we blocked caspase-8 activity in WT and TIPE2 knockdown RAW 264.7 macrophages using the caspase-8 inhibitor. By flow cytometry, we found that casapse-8 blockade significantly diminished the hyper-responsiveness of TIPE2 knockdown cells (Fig. 7D). Both the percentage of IL-6-producing cells and the mean fluorescence intensity of IL-6 were reduced. Significant reduction in IL-6 levels in the culture supernatants were also observed by ELISA (data not shown). These data indicate that TIPE2 may inhibit NF-κB activation by enzymatically active caspase-8 (Bidere et al., 2006; Su et al., 2005). In addition to activated caspase-8, the pro-domain of the caspase-8 precursor can also mediate NF-κB activation when overexpressed in vitro (Chaudhary et al., 2000; Hu et al., 2000). However, we found that TIPE2 was unable to modulate NF-κB activation induced by the caspase-8 precursor (Fig. S15).
Regulation of Fas-mediated apoptosis and activation-induced cell death by TIPE2
Because caspase-8 is an apoptosis initiator, we asked whether its new binding partner, TIPE2, regulated cell death. By comparing wild type and TIPE2 knockdown or knockout cells, we found no evidence that TIPE2 regulated cell death induced by the following agents/conditions: staurosporine, ultraviolet (UV), gamma-irradiation, etoposide, LPS, CpG, TNF-α, serum or cytokine deprivation, and cell culture conditions described in previous experiments (Figs. 7E, S16 and unpublished data). However, a role for TIPE2 in T cell death was detected under two experimental conditions. The first was the activation-induced cell death (AICD) assay in which T cells were repeatedly stimulated with anti-CD3 antibodies. Both CD4+ and CD8+ T cell death was reduced in the absence of TIPE2 (Fig. 7F). The second was the Fas ligand (FasL)-induced apoptosis of the T cell line EL-4. In the absence of FasL, wild type and TIPE2 knockdown EL-4 cells underwent similar degrees of spontaneous apoptosis. By contrast, FasL-induced apoptosis was significantly blocked in TIPE2 knockdown cells (Fig. 7G). Consistent with this result, over-expressing TIPE2 in EL-4 cells significantly enhanced FasL-induced apoptosis (Fig. S17). As discussed below, because TIPE2 is not recruited to the DISC, the reduced apoptosis in TIPE2 knockout and knockdown cells is likely secondary to its effect on cell activation.
Discussion
Results reported here indicate that TIPE2 is an essential negative regulator of inflammation and immune homeostasis. TIPE2-deficent mice suffer from chronic inflammatory diseases and TIPE2-deficient T cells and macrophages produce significantly increased levels of inflammatory cytokines. The inflammatory disease in TIPE2-deficent mice shares many similarities with those of mice and/or humans deficient in fas/fasl, caspase8, caspase10, bim, ctla4, tgfβ, il2, il10, Iκb, socs1, itch and foxp3 (Chun et al., 2002; Van Parijs and Abbas, 1998; Wahl et al., 2006). This indicates that immune homeostasis is maintained by multiple negative regulators that could not compensate for each other, because deficiency in any one of them leads to fatal inflammatory diseases. Since ~30% of genes in the murine and human genomes have not yet been studied, the total number of genes required for maintaining immune homeostasis remains to be determined.
TIPE2 in TCR and TLR signaling
Results reported here indicate that TIPE2 is a critical regulator of both TCR and TLR signaling. TCR and TLR initiate immune responses by transmitting receptor-proximal signals into immune cells through well-organized signaling complexes, or signalosomes (which consist of both common and receptor-specific signaling molecules). The receptor-proximal signals culminate in the activation of several transcription factors, which include AP-1 and NF-κB. These transcription factors in turn activate genes that mediate the activation, differentiation and effector functions of immune cells. Results reported here suggest that TIPE2 regulates TCR and TLR signaling by targeting, at least in part, signaling complexes that contain caspase-8.
Although best known as an apoptosis initiator down-stream of death receptors, caspase-8 has recently been shown to play essential roles in immune cell activation and proliferation. Caspase-8-deficiency in humans and mice not only blocks death receptor-induced apoptosis but also leads to immune deficiency (Chun et al., 2002; Salmena and Hakem, 2005). Caspase-8-deficient immune cells are defective in their responses to both TLR and antigen receptor stimulations (Bidere et al., 2006; Chun et al., 2002; Lemmers et al., 2007; Salmena and Hakem, 2005; Su et al., 2005; Takahashi et al., 2006). The effect of caspase-8 on cell activation is independent from its role in apoptosis but may require NF-κB activation mediated by the activation receptor-induced signalosome (ARIS) that contains CARMA1, Bcl10, MALT1 and IKK (Bidere et al., 2006; Lemmers et al., 2007; Su et al., 2005). Although the precise mechanism(s) through which caspase-8 activates NF-κB is not clear, two theories have been put forward. The first may be called “the pro-domain theory” which states that the pro-domain of the caspase-8 precursor recruits other components of the ARIS to activate IKK (Chaudhary et al., 2000; Hu et al., 2000). This theory is supported primarily by experiments in which caspase-8 is overexpressed. The second may be called “the active caspase theory” which states that the enzymatic activity of the caspase-8 (but not the pro-domain) is required for its effect on IKK activation (Bidere et al., 2006; Su et al., 2005). Data reported here provide additional support for the second theory and indicate that TIPE2 may regulate the active caspase-8-dependent, but not pro-domain-dependent, NF-κB activation. Additionally, because JNK and p38 phosphorylation is also enhanced in TIPE2−/− cells, but caspase-8 inhibitors block NF-κB, not MAP kinase activation, we further propose that TIPE2 may regulate the MAP kinase pathway through caspase-8-independent mechanisms.
While the CARMA1-MALT1-Bcl10-caspase-8-IKK signalosome is well characterized for T cells following TCR activation (Bidere et al., 2006; Su et al., 2005), whether a similar signaling complex forms following TLR activation is not clear. It was reported that caspase-8 recruitment to the IKK complex following TLR activation in B cells could only be detected at 30 min (Lemmers et al., 2007). Whether this occurs in macrophages is unknown. To address this issue, we performed immunoprecipitation analyses of primary murine macrophages and RAW macrophages using antibodies to caspase-8, IKKα and IKKβ We were unable to detect any caspase-8 signal using these antibodies 0–60 mins after LPS stimulation (unpublished data). These results indicate that 1) the IKK-caspase-8 complex in macrophages may be unstable, or 2) IKK and caspase-8 may reside in different signalosomes in macrophages following TLR4 activation.
TIPE2 in immune cell apoptosis and homeostasis
It is a well-recognized paradox that certain DED-containing proteins are capable of regulating both apoptosis and cell activation. These include FADD (Hua et al., 2003; Hueber et al., 2000), FLIP (Chang et al., 2002) and caspase-8 (Su et al., 2005). While the molecular mechanisms underlying this paradox are not fully understood, research about these molecules has led to the characterization of a central apoptotic pathway operating in a variety of cell types. This pathway is called extrinsic, death receptor-induced, or mitochondria-independent pathway of apoptosis. Unlike the intrinsic or mitochondria-dependent pathway that is activated primarily by ‘stress factors’ that disrupt the integrity of the outer membrane of mitochondria, the extrinsic pathway is activated by death receptors of the TNF family (Budihardjo et al., 1999). Both pathways of apoptosis are crucial for immune homeostasis and the normal functioning of the immune system. However, AICD of T cells as tested in this study relies primarily on the extrinsic pathway in which Fas plays an essential role (Kabelitz et al., 1993). Results reported here establish that TIPE2 is a novel protein involved in regulating both apoptosis and activation of T lymphocytes.
In theory, TIPE2 may regulate Fas-mediated apoptosis through two distinct mechanisms: 1) by directly modulating the activities of caspase-8 in the DISC, and 2) by altering the levels of death and survival factors that control the Fas apoptotic pathway. Because TIPE2 is not recruited to DISC upon Fas ligation (Fig. S13), the second, but not the first, mechanism is most likely operating in our system. In support of this, NF-κB activation is significantly enhanced in TIPE2 knockdown cells both under resting and activating conditions (Figs. 7A, S9B and S10). Because NF-κB is an effective inhibitor of Fas-induced apoptosis (Dudley et al., 1999; Rivera-Walsh et al., 2000), we propose that the increased NF-κB activation is responsible for the resistance of TIPE2-deficient cells to Fas-induced apoptosis. With the exception of the DED-like domain, TIPE2 and the other three members of its family do not share sequence homology with other DED-containing proteins or any other known proteins. Therefore, the TIPE2 family may represent a novel class of apoptosis regulators.
Our data indicate that TIPE2 knockout T cells do not have an intrinsic defect in proliferation when activated in vitro with anti-CD3 and anti-CD28 (Fig. 4D). Therefore, splenomegaly and leukocytosis in TIPE2−/− mice are likely mediated by proliferation-independent mechanisms. These may include 1) resistance of immune cells to activation-induced cell death as shown in Fig. 7, and 2) chronic expansion of immune cells in vivo as a result of increased production of cytokines that promote proliferation.
In summary, we have isolated a novel gene from inflamed nervous tissue and discovered that it is an essential negative regulator of both innate and adaptive immunity. Understanding the basic biology of this new molecule should provide novel insights into the mechanisms of inflammatory diseases and immune homeostasis.
Experimental Procedures
Cloning of TIPE2
An expressed sequence tag (AI847688) was identified from a microarray analysis (Carmody et al., 2002) as one of the 141 genes preferentially expressed in inflamed but not normal spinal cord. A putative mouse full-length sequence was generated by aligning the EST sequence with the mouse EST database using the BLAST program. This gave rise to a novel cDNA of 555 bp (Gene bank accession number AF548004). Full-length TIPE2 was then isolated from the spinal cord of mice with EAE by RT-PCR using TIPE2-specific primers. A human testis cDNA clone that bears a high degree of homology with mouse TIPE2 was identified by BLAST search of the human EST database, and purchased directly from ATCC (gene bank accession number BG258888, clone number 4509736). The full-length TIPE2 was then sequenced and cloned into the expression vector pRK5 with FLAG or HA tag at the C-terminus.
Northern blot
Total RNA of cells or tissues was prepared using the Trizol reagent (GIBCOL BRL, Carlsbad, CA), fractionated by electrophoresis, transferred to a nylon membrane and hybridized at 60°C with 32P-labeled DNA probes specific for TIPE2 or GAPDH.
Generation of TIPE2-deficient mice
A 2.2 kb and a 5.0 kb TIPE2 genomic fragments were amplified by PCR and cloned into the Xhol/Nhe I and the Not I/Sal I sites of the pOSDUPDEL vector (a gift from Dr. X-P Zhong, Duke University), respectively. TL1 embryonic stem cells from 129S6/SvEvTac mice were transfected with the targeting vector and subjected to positive and negative selection using G418 and ganciclovir, respectively. Two ES cell clones were identified by Southern blot, which had a copy of the TIPE2 gene (including both exons 1 and 2) replaced by the neomycin resistance gene cassette. The mutant ES cells were injected into 4-day-old C57BL/6J mouse blastocysts. The resultant chimeric male offsprings were crossed with 129S6/SvEvTac females for germline transmission. Unless indicated otherwise, all mice used in this study were of the 129S6/SvEvTac background. Age and sex matched littermates were used as controls. Mice were housed in the University of Pennsylvania Animal Care Facilities under pathogen-free conditions. All procedures used were pre-approved by the Institutional Animal Care and Use Committee.
T cell cytokine and proliferation assays
Single cell suspensions of the lymph nodes and spleens were depleted of red blood cells using a hemolysis buffer. CD4+ T cells were then purified using the CD4 (L3T4) MicroBeads (Miltenyi Biotec) according to the manufacturer’s protocol. Cells were cultured at a density of 1×106 per well in U-bottom 96-well plates in the presence of various concentrations of plate-bound anti-CD3 mAb and 2 μg/ml soluble anti-CD28 mAb. After 48 hrs, the culture supernatants were collected and cytokine concentrations determined by ELISA. For the proliferation assay, cells were cultured at 0.5×106 per well in the presence of the same antibodies described above, and pulsed with 3H-thymidine (1μCi/well) at 40 hr. Radioactivity was measured using a flat-bed beta counter (Beckman).
Activation-induced cell death (AICD) and cytokine withdrawal-induced cell death
Splenocytes, 3×106/well, were cultured for 48 hrs in complete DMEM containing 20 U/ml IL-2 and 1 μg/ml anti-CD28 mAb in 48-well plates precoated with 1 μg/ml anti-CD3 mAb. For AICD assay, live cells were purified through a ficoll density gradient and re-stimulated with plate-bound anti-CD3 mAb (10 μg/ml) for 24 hrs. For cytokine withdrawal-induced cell death assay, live cells were cultured for 24 hrs in complete DMEM containing no antibodies or exogenous cytokines. Cells were then stained with 7-AAD, annexin V and antibodies to CD4 and CD8. The percentage of apoptotic cells was determined by flow cytometry.
Flow cytometry
For cell surface markers, cells were stained with antibodies in PBS with 2% BSA and 0.05% sodium azide for 20 min at 4°C. For intracellular cytokines, cells were treated with Cytofix/Cytoperm kit and stained with anti-cytokine antibodies (BD Biosciences). After washing, cells were analyzed on a four-color FACS Calibur (BD Biosciences). Data analysis was performed using the CellQuest software.
TIPE2 knockdown
TIPE2 siRNA (5′-GAAGTGAAACTCAGGTCCG-3′) or scrambled control siRNA (5′-AGTGAAACAGTGCAGCTGC-3′) was cloned into the lentiviral vector pLL3.7 that carries the green fluorescent protein (GFP) cDNA. Lentiviruses were produced by co-transfecting 293 T cells with the pLL3.7 plasmids and packaging vectors (Rubinson et al., 2003). To knockdown TIPE2 expression, RAW 264.7 or EL-4 cells were infected with recombinant lentiviruses that carry the TIPE2 siRNA. Cells infected with the same numbers of recombinant lentiviruses that carry the control siRNA serve as wild type controls. GFP was used to track virus-infected cells. For the RAW 264.7 line, infected cells constitute more than 90% of the total, whereas for the EL-4 line, infected cells constitute ~10% of the total. Analysis was performed on gated GFP+ cells for the EL-4 line.
Statistical analyses
The differences in protein and mRNA levels were analyzed by Student’s t test. The differences in survival rates were analyzed by Mann-Whitney U test.
Supplementary Material
Acknowledgments
The authors thank Drs. Wenchao Song, Warren Pear and Xiaoping Zhong for advices and reagents, Liudmila Mazaleuskaya, Chia-Ying Yang, Joanna DiSpirito, Jean Richa and members of the transgenic and morphological cores for technical supports. Supported by grants from the National Institutes of Health (AI50059, DK070691 and AI069289), USA.
Footnotes
Author contributions.
S. Gong generated TIPE2 knockdown and overexpressing cells, and performed the LCMV and septic shock experiments in KO mice and most of the signaling studies. H. Sun cloned the TIPE2 gene, generated TIPE2 knockout mice and carried out characterizations of the mice. YHC supervised the project and wrote the paper. Others contributed to the generation of various figures in the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bidere N, Snow AL, Sakai K, Zheng L, Lenardo MJ. Caspase-8 regulation by direct interaction with TRAF6 in T cell receptor-induced NF-kappaB activation. Current Biology. 2006;16:1666–1671. doi: 10.1016/j.cub.2006.06.062. [DOI] [PubMed] [Google Scholar]
- Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annual Review of Cell & Developmental Biology. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- Carmody R, Hilliard B, Chodosh L, Chen Y. Genomic scale profiling of autoimmune inflammation in the central nervous system: the nervous response to inflammation. Journal of Neuroimmunology. 2002;133:95–107. doi: 10.1016/s0165-5728(02)00366-1. [DOI] [PubMed] [Google Scholar]
- Chang DW, Xing Z, Pan Y, Algeciras-Schimnich A, Barnhart BC, Yaish-Ohad S, Peter ME, Yang X. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO Journal. 2002;21:3704–3714. doi: 10.1093/emboj/cdf356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary PM, Eby MT, Jasmin A, Hood L. Activation of the c-Jun N-terminal kinase/stress-activated protein kinase pathway by overexpression of caspase-8 and its homologs. Journal of Biological Chemistry. 1999;274:19211–19219. doi: 10.1074/jbc.274.27.19211. [DOI] [PubMed] [Google Scholar]
- Chaudhary PM, Eby MT, Jasmin A, Kumar A, Liu L, Hood L. Activation of the NF-kappaB pathway by caspase 8 and its homologs. Oncogene. 2000;19:4451–4460. doi: 10.1038/sj.onc.1203812. [DOI] [PubMed] [Google Scholar]
- Chun HJ, Zheng L, Ahmad M, Wang J, Speirs CK, Siegel RM, Dale JK, Puck J, Davis J, Hall CG, et al. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature. 2002;419:395–399. doi: 10.1038/nature01063. [DOI] [PubMed] [Google Scholar]
- Combet C, Blanchet C, Geourjon C, Deleage G. NPS@: network protein sequence analysis. Trends in Biochemical Sciences. 2000;25:147–150. doi: 10.1016/s0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- Dudley E, Hornung F, Zheng L, Scherer D, Ballard D, Lenardo M. NF-kappaB regulates Fas/APO-1/CD95- and TCR- mediated apoptosis of T lymphocytes. European Journal of Immunology. 1999;29:878–886. doi: 10.1002/(SICI)1521-4141(199903)29:03<878::AID-IMMU878>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Hu WH, Johnson H, Shu HB. Activation of NF-kappaB by FADD, Casper, and caspase-8. Journal of Biological Chemistry. 2000;275:10838–10844. doi: 10.1074/jbc.275.15.10838. [DOI] [PubMed] [Google Scholar]
- Hua ZC, Sohn SJ, Kang C, Cado D, Winoto A. A function of Fas-associated death domain protein in cell cycle progression localized to a single amino acid at its C-terminal region. Immunity. 2003;18:513–521. doi: 10.1016/s1074-7613(03)00083-9. [DOI] [PubMed] [Google Scholar]
- Hueber AO, Zornig M, Bernard AM, Chautan M, Evan G. A dominant negative Fas-associated death domain protein mutant inhibits proliferation and leads to impaired calcium mobilization in both T-cells and fibroblasts. Journal of Biological Chemistry. 2000;275:10453–10462. doi: 10.1074/jbc.275.14.10453. [DOI] [PubMed] [Google Scholar]
- Kabelitz D, Pohl T, Pechhold K. Activation-induced cell death (apoptosis) of mature peripheral T lymphocytes [Review] Immunology Today. 1993;14:338–339. doi: 10.1016/0167-5699(93)90231-9. [DOI] [PubMed] [Google Scholar]
- Kumar D, Gokhale P, Broustas C, Chakravarty D, Ahmad I, Kasid U. Expression of SCC-S2, an antiapoptotic molecule, correlates with enhanced proliferation and tumorigenicity of MDA-MB 435 cells. Oncogene. 2004;23:612–616. doi: 10.1038/sj.onc.1207123. [DOI] [PubMed] [Google Scholar]
- Kumar D, Whiteside TL, Kasid U. Identification of a novel tumor necrosis factor-alpha-inducible gene, SCC-S2, containing the consensus sequence of a death effector domain of fas-associated death domain-like interleukin- 1beta-converting enzyme-inhibitory protein. Journal of Biological Chemistry. 2000;275:2973–2978. doi: 10.1074/jbc.275.4.2973. [DOI] [PubMed] [Google Scholar]
- Lemmers B, Salmena L, Bidere N, Su H, Matysiak-Zablocki E, Murakami K, Ohashi PS, Jurisicova A, Lenardo M, Hakem R, Hakem A. Essential role for caspase-8 in Toll-like receptors and NFkappaB signaling. Journal of Biological Chemistry. 2007;282:7416–7423. doi: 10.1074/jbc.M606721200. [DOI] [PubMed] [Google Scholar]
- Nagata S, Suda T. Fas and Fas ligand: lpr and gld mutations. Immunology Today. 1995;16:39–43. doi: 10.1016/0167-5699(95)80069-7. [DOI] [PubMed] [Google Scholar]
- Ochsenbein AF, Karrer U, Klenerman P, Althage A, Ciurea A, Shen H, Miller JF, Whitton JL, Hengartner H, Zinkernagel RM. A comparison of T cell memory against the same antigen induced by virus versus intracellular bacteria. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:9293–9298. doi: 10.1073/pnas.96.16.9293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Wang FH, Whiteside TL, Kasid U. Identification of seven differentially displayed transcripts in human primary and matched metastatic head and neck squamous cell carcinoma cell lines: implications in metastasis and/or radiation response. Oral Oncology. 1997;33:197–203. doi: 10.1016/s0964-1955(96)00065-6. [DOI] [PubMed] [Google Scholar]
- Rivera-Walsh I, Cvijic ME, Xiao G, Sun SC. The NF-kappa B signaling pathway is not required for Fas ligand gene induction but mediates protection from activation-induced cell death. Journal of Biological Chemistry. 2000;275:25222–25230. doi: 10.1074/jbc.M000444200. [DOI] [PubMed] [Google Scholar]
- Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Ihrig MM, McManus MT, Gertler FB, et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nature Genetics. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- Salmena L, Hakem R. Caspase-8 deficiency in T cells leads to a lethal lymphoinfiltrative immune disorder. Journal of Experimental Medicine. 2005;202:727–732. doi: 10.1084/jem.20050683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K, Chen Y, Goke R, Wilmen A, Seidel C, Goke A, Hilliard B, Chen Y. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is an inhibitor of autoimmune inflammation and cell cycle progression. J Exp Med. 2000;191:1095–1104. doi: 10.1084/jem.191.7.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Bidere N, Zheng L, Cubre A, Sakai K, Dale J, Salmena L, Hakem R, Straus S, Lenardo M. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science. 2005;307:1465–1468. doi: 10.1126/science.1104765. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Kawai T, Kumar H, Sato S, Yonehara S, Akira S. Roles of caspase-8 and caspase-10 in innate immune responses to double-stranded RNA. Journal of Immunology. 2006;176:4520–4524. doi: 10.4049/jimmunol.176.8.4520. [DOI] [PubMed] [Google Scholar]
- Van Parijs L, Abbas AK. Homeostasis and self-tolerance in the immune system: turning lymphocytes off. Science. 1998;280:243–248. doi: 10.1126/science.280.5361.243. [DOI] [PubMed] [Google Scholar]
- Wahl SM, Wen J, Moutsopoulos N. TGF-beta: a mobile purveyor of immune privilege. Immunological Reviews. 2006;213:213–227. doi: 10.1111/j.1600-065X.2006.00437.x. [DOI] [PubMed] [Google Scholar]
- Yoshinaga-Hirabayashi T, Yoneda Y. Expression of SCC in ovarian granulosa cells and cultured cells, induced rapid structural changes in mitochondria. Italian Journal of Anatomy & Embryology. 2001;106:51–57. [PubMed] [Google Scholar]
- Zhang C, Chakravarty D, Sakabe I, Mewani RR, Boudreau HE, Kumar D, Ahmad I, Kasid UN. Role of SCC-S2 in experimental metastasis and modulation of VEGFR-2, MMP-1, and MMP-9 expression. Molecular Therapy: the Journal of the American Society of Gene Therapy. 2006;13:947–955. doi: 10.1016/j.ymthe.2005.11.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.