

Abstract
The Escherichia coli iron transporter, FepA, has a globular N terminus that resides within a transmembrane β-barrel formed by its C terminus. We engineered 25 cysteine substitution mutations at different locations in FepA and modified their sulfhydryl side chains with fluorescein maleimide in live cells. The reactivity of the Cys residues changed, sometimes dramatically, during the transport of ferric enterobactin, the natural ligand of FepA. Patterns of Cys susceptibility reflected energy- and TonB-dependent motion in the receptor protein. During transport, a residue on the normally buried surface of the N-domain was labeled by fluorescein maleimide in the periplasm, providing evidence that the transport process involves expulsion of the globular domain from the β-barrel. Porin deficiency much reduced the fluoresceination of this site, confirming the periplasmic labeling route. These data support the previously proposed, but never demonstrated, ball-and-chain theory of membrane transport. Functional complementation between a separately expressed N terminus and C-terminal β-barrel domain confirmed the feasibility of this mechanism.
FepA is a Gram-negative bacterial outer membrane (OM)6 protein that transports ferric enterobactin (FeEnt) (1-3). The crystal structures of FepA (4) and other bacterial metal transporters (FhuA, BtuB, and FpvA (15-17, 57)), contain a C-terminal, 22-stranded β-barrel, placing them in the porin super-family (5). Their ∼150-residue globular N termini (N-domain; see Fig. 1) reside within their β-barrels. This architecture is potentially consistent with the “ball-and-chain” mechanism of membrane transport, whereby the globule controls solute (ligand) uptake by moving in and out of the channel. This process was postulated for nervous system channels (6), but no demonstrated examples of ball-and-chain transport are known.
FIGURE 1.
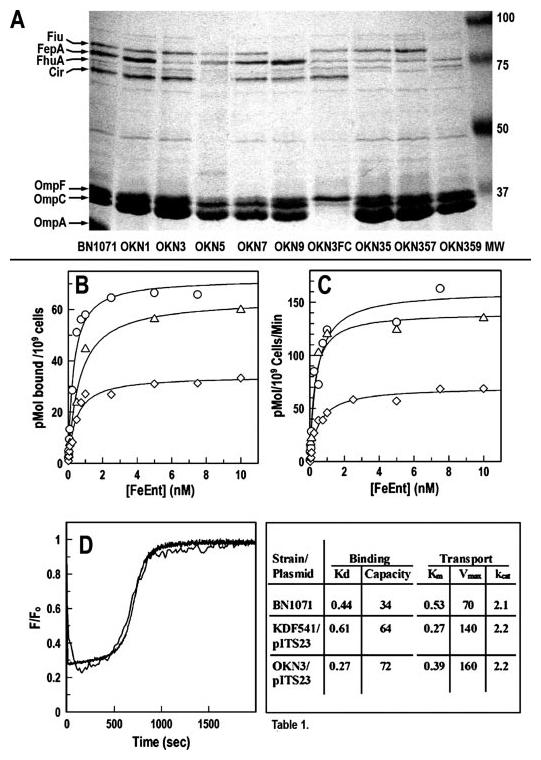
A, outer membranes of E. coli strains with site-directed deletions of siderophore receptor proteins. We genetically engineered (36) E. coli K12 strain BN1071 (entA (35)) to introduce individual deletions of tonB, fecA, fepA, cir, fhuA, and ybiL (strains OKN1, -2, -3, -5, -7, and -9, respectively). In addition, we transduced the defective ompF::Kn and ompC::Tc genes from HN705 (49) into OKN3 (OKN3FC) and generated multiple combinations of the individual deletions (OKN35: ΔfepA, Δcir; OKN357: ΔfepA, Δcir, ΔfhuA; OKN359: ΔfepA, Δcir, ΔybiL; and OKN2357: ΔfecA, ΔfepA, Δcir, ΔfhuA). Sarkosyl-extracted OM fractions of selected strains, which were grown in MOPS media, show the identity of the siderophore receptor proteins and their absence in the mutant bacteria. B and C, 59FeEnt binding and transport by OKN3/pITS23. After transformation of OKN3 with pITS23 (fepA+, ○), we determined its ability to bind (B) and transport (C) 59FeEnt, relative to BN1071 (chromosomal fepA+, ◇) and KDF541/pITS23 ((38), Δ). The latter host strain (48) carries a spontaneous deletion of the C-terminal 381 amino acids of FepA (45). D, fluorescence spectroscopic measurements of FeEnt uptake. After modification with FM, we compared FeEnt uptake (47) by OKN3/pFepAS271C (thin line) and KDF541/pFepAS271C (heavy line; data from Ref. 47). The fluorescence spectroscopic kinetic curves were superimposable. The KDF541 plot is an average of three trials, whereas that of OKN3 is from a single experiment. See Ref. 47 for further details. The table shows a summary of 59FeEnt binding and transport by host strains. Aside from higher expression levels conferred by the plasmid systems, the thermodynamic and kinetic parameters of chromosomal- and plasmid-derived FeEnt transport systems (Kd and Km (nm), capacity (pmol/109 cells), Vmax (pmol/109 cells/min), kcat (expressed as turnover number, molecules transported/receptor/min)) were equivalent in the three strains.
FepA and its relatives are unlike other porins (7, 8), because they selectively adsorb metal chelates with high affinity (3, 9-14). Ligand binding causes small conformational changes that activate them to transport competency (15-17), hence their designation “ligand-gated porin” (LGP). The requirements for metabolic energy (18-20) and another cell envelope protein, TonB (21-24), in LGP mediated transport are well known but unaccounted for: the OM has no source of energy and cannot sustain an ion gradient because of its open porin channels (7); TonB is a minor cell envelope protein whose functions are not yet understood.
In live cells, FepA binds and transports FeEnt via sub-reactions with different dependences on energy and TonB. (i) In the absence of ligand the receptor opens, and its flexible surface loops extend outward (25). (ii) FeEnt binds to FepA in a biphasic reaction (26) that begins with adsorption to aromatic amino acids in the loop extremities (27, 28). Multiple determinants in multiple loops, including L7 (25), converge on the iron complex, creating a closed conformation that associates the negatively charged (−3), catecholate iron center with basic and aromatic residues in the receptor's vestibule (27, 29, 30). At binding equilibrium FeEnt sits atop the N-domain, poised for transport through the β-barrel. (iii) In the paralog, FhuA, ferrichrome binding relocates a short β-strand called the TonB-box away from the β-barrel wall, sending a signal of receptor occupancy from the cell surface to the periplasm (15, 16). No data exist on whether the same phenomenon takes place when FepA binds FeEnt, but conformational motion also occurs in the TonB-box of BtuB when it binds cyanocobalamin (31). These initial stages of ligand uptake are energy- and TonB-independent, because they happen with equivalent affinity and rate in energy-sufficient or -deficient, and TonB+ or tonB cells. (iv) In energized tonB+ cells FepA internalizes FeEnt through its transmembrane channel into the periplasm. Transport does not occur in de-energized or tonB cells.
The function of the N-domain in transport is presently inscrutable (32). It has low affinity for the ligand (32), and it blocks the FepA channel, necessitating a structural rearrangement to open a pathway to the periplasm. In the ball-and-chain model the N-domain dislodges into the periplasm, either as a globule that moves on the basis of its hinge-like connection to the β-barrel (at residue 150), or by unfolding of its α-β structure, in both cases opening the large (∼40-Å diameter) trans-OM channel. N-domain expulsion rationalizes ligand internalization: if the metal complex associates with the N-domain loops, then movement of the N-domain to the periplasm will transport FeEnt through the channel.
The Transient Pore model, on the other hand, requires conformational motion in the N-domain while it is resident in the β-barrel, which opens a necessarily smaller pore for passage of the metal complex. The dimensions of FeEnt indicate that such a channel must acquire a diameter of ∼20 Å. According to this mechanism, the ligand transits the protein interior by passing between sequential binding sites with affinity for its natural chemistry. For the acidic siderophore FeEnt, this may happen by stepwise binding to basic residues that exist near the top of the FepA N-domain and line its membrane channel. In this report we tested for the main difference between these models, N-domain movement into the periplasm, by determining the susceptibility of genetically engineered Cys residues to modification by fluorescein maleimide (FM) during FeEnt uptake.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, Culture Conditions, and Chemicals
Bacteria were grown at 37 °C with vigorous shaking in Luria-Bertani (LB) broth (33) containing streptomycin (100 μg/ml) and chloramphenicol (20 μg/ml). For physiological and biochemical experiments, including fluorescence labeling studies, we subcultured the bacteria (1%) from stationary-phase LB broth cultures into MOPS minimal medium (58) or T-medium (2) containing the same antibiotics but without added iron. After ∼6 h the growth rate diminished from iron deficiency. Under these conditions the expression of the inner membrane (FepC, FepD, and FepG) and periplasmic (FepB) components of the transport system reached maximum levels, and transport of FeEnt through the OM is the rate-limiting step in its uptake (30, 34).
Siderophores and Colicin B
We formed iron complexes of purified enterobactin (28) and TRENCAM (a gift from Dick van der Helm) by dissolving the siderophores in methanol, incubating them with an equimolar amount of freshly prepared FeSO4 in water, or 59FeCl3 (Amersham Biosciences) in dilute HCl. After 1 h at 25°C we adjusted the pH to 6.9 with NaHPO4, chromatographically purified the ferric siderophores over Sephadex LH20 in 5 mm NaHPO4, and spectrophotometrically determined their concentrations from their extinction maxima (5.6 mm−1) at 495 nm. We purified colicins B and D from cultures of Escherichia coli strains DM1187/pCLB1 (26) and CA23, respectively.
Site-directed Deletion Mutagenesis
We genetically engineered in-frame chromosomal deletion mutants in E. coli K-12 strain BN1071 (35) by allelic replacement (36). After verification of OKN1 (ΔtonB), OKN2 (ΔfecA), OKN3 (ΔfepA), OKN5 (Δcir), OKN7 (ΔfhuA), OKN9 (Δfiu), and OKN13 (ΔtonB and ΔfepA) by PCR reactions from chromosomal DNA and SDS-PAGE analysis of their cell envelopes (Fig. 1), we transformed OKN3 and OKN13 with plasmids of interest and confirmed their expected phenotypes by measurement of FeEnt and Fc transport, and colicin and bacteriophage susceptibility (30). We constructed OKN7 by P1 transduction of the ΔfhuA marker in strain MB97 (37), which was a gift from V. Braun.
Site-directed Cys Substitution Mutagenesis
We engineered mutations on pITS23 (25, 38), a derivative of the low copy vector pHSG575 (39) carrying fepA under its natural promoter. Using QuikChange (Stratagene) we generated site-directed Cys substitution mutations in fepA on pITS23 for the following residues: Ile-14, Ala-33, Gly-54, Ser-92, Thr-107, Thr-216, Ser-254, Ala-261, Ser-271, Gly-300, Ala-322, Thr-367, Ala-383, Ser-411, Ser-470, Ser-482, Ser-507, Thr-550, Gly-565, Ser-569, Thr-585, Ser-600, Thr-609, Thr-666, and Ala-698. After verification by DNA sequence analysis (McLab, San Francisco, CA), we evaluated the colicin B and D sensitivity, ability to transport FeEnt, and the expression of the mutant FepA proteins (30): their phenotypes were indistinguishable from wild-type FepA.
Membrane Separations
Bacterial cells, grown to late-exponential phase in MOPS minimal media, were collected by centrifugation, suspended in either 0.01 m HEPES, pH 7.4, or 0.01 m Tris-Cl, pH 7.4, 0.9% NaCl (Tris-buffered saline (TBS)), chilled on ice, and lysed by passage through a French pressure cell at 14,000 p.s.i. The OM and inner membrane (IM) fragments formed by French press lysis (40) were either fractionated by sucrose gradient centrifugation (41) or subjected to differential extraction with sodium sarkosinate (42).
SDS-PAGE, Western Immunoblots, and Expression Measurements
For SDS-PAGE, samples were suspended in SDS-containing sample buffer plus 3% β-mercaptoethanol, boiled for 5 min, and electrophoresed (30, 43) at room temperature. For Western immunoblots, proteins were transferred to nitrocellu-lose paper, the paper was blocked with 50 mm Tris chloride (pH 7.5), 0.5 m NaCl (TBS) plus 1% gelatin, incubated with mouse anti-FepA monoclonal antibody 45 (0.5% (44)) in TBS plus 1% gelatin for 3 h, and developed with 125I-protein A (30).
For determinations of protein expression levels, 5 × 108 bacteria were collected by centrifugation, resuspended in 150 μl of SDS-PAGE sample buffer, boiled for 5 min, briefly centrifuged to remove debris, and 108 cells were subjected to SDS-PAGE and Western immunoblot. After overnight exposure on an imaging screen, radioactivity was quantitated on the STORM-SCAN PhosphorImager.
Fluorescence Labeling
For covalent modification of Cys residues in live cells, after overnight growth in LB broth the bacteria were subcultured in MOPS minimal media and shaken at 37 °C until they reached mid-log phase (4–5 × 108 cells/ml; ∼5 h). The cells were collected by centrifugation, washed twice by suspension in TBS (pH 7.4, 0 °C) and centrifugation, and then resuspended in TBS (pH 7.2, 0 °C) containing glucose (0.4%), with or without FeEnt (0.0001–200 μm) or sodium azide (10 mm), as the experiments warranted. FM was prepared by dissolution in dimethyl formamide; its concentration was spectrophotometrically determined (∊493 nm = 81,500 m−1) after dilution into 10 mm Tris-Cl, pH 8. FM was added to 5–20 μm, for 1–15 min at 37 °C in the dark. The reactions were quenched by the addition of cysteine (to 1 mm), and the bacteria were washed three times with 1 ml of ice-cold TBS, resuspended in ice-cold TBS, quantitated by visible spectrophotometry at 600 nm, and lysed by boiling in SDS-PAGE sample buffer. Lysates were subjected to SDS-PAGE, and the gels were analyzed by direct fluorescence imagery.
Fluorescence Imagery
Immediately following resolution of cell lysate proteins by SDS-PAGE, the slab gels were rinsed with water and transferred to a STORMSCAN PhosphorImager and scanned for fluorescence emissions, and the resultant images were analyzed by ImageQuant 5.2 (Molecular Dynamics). FepA (cysteine-substituted) was the primary protein that FM labeled in the outer membranes, but other proteins in the lysates were also consistently modified. The intensity of FepA Cys mutant fluoresceination was quantitated relative to the total labeling that was observed for all other proteins in the cell lysate.
Disulfide Bonds between the N-domain and the β-Barrel
Bacteria were cultured in LB broth and subcultured in nutrient broth containing β-mercaptoethanol, dithiothreitol, or oxidized dithiothreitol at the indicated concentrations. After 3.5 h aliquots of bacteria were collected by centrifugation, lysed, and analyzed by SDS-PAGE and Western immunoblot with anti-FepA monoclonal antibody 45, or subjected to siderophore nutrition tests.
Complementation Assays
E. coli strain OKN3, which carries a precise deletion of the fepA structural gene (see “Results”), was individually transformed with plasmids expressing the N-domain (pFepN) and empty β-barrel (pFepβ) of FepA and with both plasmids (pFepNpFepβ). We verified the expression of the individual domains by Western immunoblot and then determined the functionality of the two protein domains when expressed singly or together, by measuring susceptibility to colicin B and D, and by FeEnt uptake.
RESULTS
Precise Deletions of OM Receptor Proteins
Because of potential complementation between chromosomal fragments of ferric siderophore receptor genes and mutant receptor genes on plasmids (45), we genetically engineered (36) an isogenic series of bacterial strains containing precise deletions of tonB, fepA, fepA, cir, fhuA, and fiu in BN1071 (F−trp, entA−). After transformation of OKN3 (ΔfepA) with pITS23 (fepA+ (38)), and OKN7 (ΔfhuA) with pITS11 (fhuA+ (38)), we tested all the strains for their susceptibility to bacteriocins B, D, and M, bacteriophages T5 and H87, and for their ability to bind and transport ferric enterobactin and ferrichrome. The resistance of the hosts strains to the colicins and phage showed the absence of FepA and FhuA, and the complementation of the engineered deletion strains to sensitivity by fepA+ and fhuA+ alleles on pHSG575 verified this conclusion. Similarly, OKN3 and OKN7 were unable to transport ferric enterobactin and ferrichrome, respectively, whereas the thermodynamic and kinetic parameters of OKN3/pITS23 and OKN7/pITS11 were identical to those of wild-type FepA and wild-type FhuA in KDF541 harboring the same plasmids, and in BN1071, which expresses the wild-type proteins from the chromosome (25, 27, 28, 30, 38). Lastly, fluorescence spectroscopic measurements of FeEnt uptake in FM-modified OKN3/FepAS271C (Fig. 1) were indistinguishable from those observed in KDF541 (47). Thus, when harboring plasmids that encode the wild-type fepA and fhuA genes, host strains with precise deletions of siderophore receptor genes behaved identically to previously isolated spontaneous fepA and fhuA mutants (48).
Besides the site-directed deletions of individual siderophore receptor genes in BN1071(ΔfepA, ΔfhuA, ΔybiL (Δfiu), ΔfecA, and Δcir; Fig. 1), we constructed a derivative with an exact deletion of tonB (OKN1), another that carried inactivating transposon cassettes in ompF and ompC (from HN705 (49)) and several multiple combinations of these mutations (Fig. 1).
Modification of Genetically Engineered Cys Residues by FM
After generating 25 Cys substitutions, we confirmed the functionality of the mutant FepA proteins in activity assays. All the Cys mutant derivatives of FepA transported FeEnt like the wild-type parent protein in siderophore nutrition tests and conferred normal susceptibility to colicin B. The mutant proteins, under control of the wild-type FepA promoter, were expressed at wild-type levels (Fig. 2). Hence, in general, the introduction of a single, unpaired cysteine did not impair the physiological functions of the outer membrane protein. We surveyed the sulfhydryl side chains for their accessibility to FM (Fig. 2). When exposed for 15 min at 37 °C, with or without FeEnt, several residues were differentially reactive. The fluorophore strongly labeled engineered Cys residues in surface loops (at positions 216, 271, 322, 383, 482, and 550), reacted at lower levels with sulfhydryls in the periplasm (residues 14, 33, 300, and 666), and importantly, modified a Cys residue in the protein interior on the surface of the N-domain (residue 54). The intensity of labeling at some of these sites changed in response to the binding and transport of FeEnt (residues 14, 54, 322, 383, and 550), reiterating the receptor's dynamic response to interactions with its ligand. It was of interest that FM labeled residue G54C, buried in the receptor's interior on the surface of the N-domain, but did not label contiguous residues on the wall of the β-barrel (G565C, S569C, and T585C). The modification of G54C was weaker than that of surface residues but, nevertheless, distinct and reproducible.
FIGURE 2. FM labeling of genetically engineered Cys residues in FepA.
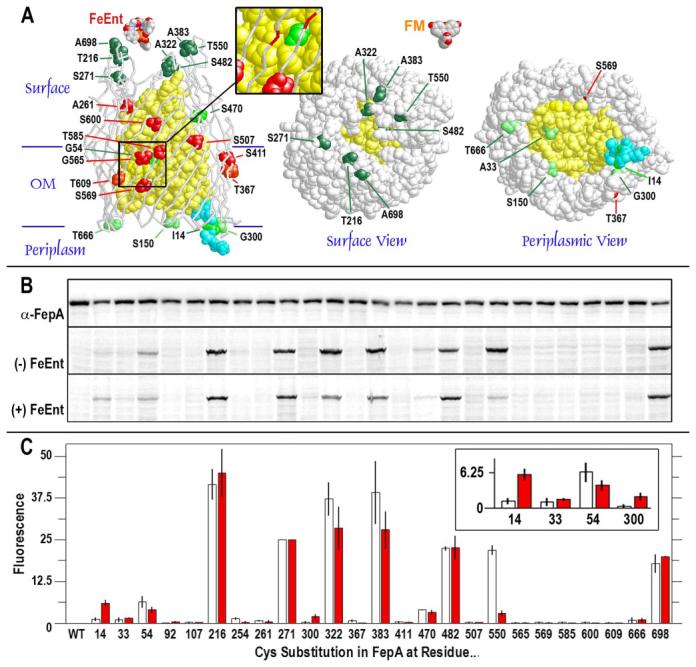
A, the N-domain is yellow, the C-domain is white, and the TonB-box (amino acids 11–17) is cyan. Left, the positions of 25 Cys substitutions of interest for fluoresceination are shown in space-filling format on a backbone representation of FepA tertiary structure. Reactive residues are colored dark green, green, and light green in relation to the intensity of their fluoresceination, as determined in B and C, below. Unreactive Cys substitutions are red. The inset shows a magnification of the region near residues G54C on the N-domain surface, opposite G565C, S569C, and T585C, whose side chains project inward from the wall of the β-barrel. Center and right, the space-filling models show the apparent accessibility, from x-ray data (4), of Cys substitutions to extrinsic fluors from the cell surface and periplasm, respectively, and the relative sizes of FeEnt and fluorescein. B and C, expression and FM modification of FepA Cys substitution mutants. B (top panel), when visualized by anti-FepA (monoclonal antibody 45 (44)) immunoblots, developed with [125I]protein A (30), the expression of wild-type (WT) and mutant FepA proteins (enumerated) was invariant. Conversely, fluorescence scans of lysates from cells expressing single Cys mutants showed that FM differentially labeled the various sulfhydryl groups in the absence (center panel) or presence (bottom panel) of FeEnt. C, quantitation of imaging data from electropherograms of SDS-PAGE gels. The bar graph depicts the intensities of Cys residue labeling (as a percentage of total cellular labeling; mean of two experiments) and the standard error of the means (vertical bars) in the absence (white) or presence (red) of FeEnt.
Cellular Distribution of Fluoresceinated Proteins
When live E. coli was exposed to FM, besides the Cys substitution mutants of FepA many other cellular proteins were weakly modified by the reagent, and about seven proteins were reproducibly labeled at higher levels (Fig. 3). The extent of labeling of other proteins depended on two factors: the external concentration of FM and the duration of the labeling reaction. Results on the concentration and time dependence of fluoresceination (data not shown) led us to employ FM at 5 μm, for 15 min at 37 °C in TBS, pH 7.2, as standard conditions. This protocol yielded specific labeling of the FepA Cys mutants (i.e. without modification of wild-type FepA) and background labeling of other proteins at moderate levels. The intensity of Cys reactivity at different sites in FepA was as high as 45% of total cellular labeling (S216C (Fig. 2)). A residue of prior interest, S271C (47), was consistently labeled at a level of 26% of total cellular labeling, in the presence or absence of FeEnt, and we used the extent of S271C fluoresceination as a control standard to which we compared the labeling at other sites.
FIGURE 3. Fractionation of fluoresceinated bacteria.
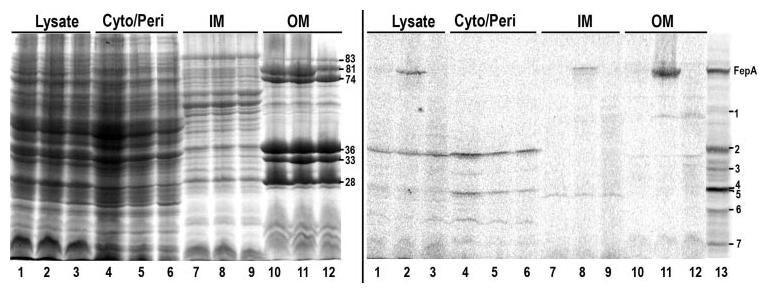
OKN3 (fepA) harboring pITS23 (fepA+; lanes 1, 4, 7, and 10) or pFepAG54C (lanes 2, 5, 8, and 11) and OKN13 (fepA, tonB)/pITS23 (lanes 3, 6, 9, and 12) were grown in MOPS media to late log, pelleted by centrifugation and lysed in a French press. Their cytoplasm/periplasm (cyto/peri), IM and OM fractions were collected, resolved by SDS-PAGE, scanned for fluorescence (right panel), and the same gel was then stained with Coomassie Blue (left panel). The molecular weights of the outer membrane proteins Fiu (83 kDa), FepA (81 kDa), CirA (74 kDa), OmpF/C (36 kDa), and OmpA (denatured (33 kDa) and native 28 kDa)) are shown (respectively, top to bottom). Lane 13 shows a fluoresceinated lysate of OKN3/pFepAG54C, without any fractionation.
G54C was one residue whose susceptibility to FM changed during the binding and transport of FeEnt, and we characterized the overall pattern of its fluorescent modification in bacteria expressing FepA with this substitution mutation. Fractionation of fluoresceinated OKN3/pFepAG54C showed the bulk of the FepAG54C-FM in the OM (∼90%) and a small amount of unprocessed, pre-FepAG54C in the IM (Fig. 3). In addition, among the approximately seven other cellular proteins that FM reacted with (seen in cell lysates, Fig. 3, lane 13), four or five resided in the periplasm/cytoplasm (Fig. 3, bands 2, 3, 4, 6, and 7; the fractionation procedure did not differentiate between the two compartments, which were collected together as the supernatant from centrifugation of the cell lysate), one derived from the IM (band 5), and one inhabited the OM (band 1). Despite these side reactions, residue G54C in FepA, which was one of most weakly labeled sites that we considered (at levels between 5 and 9% of total cellular labeling; Figs. 2, 4, and 5), was still the predominant OM target that FM reacted with, constituting >90% of the total fluorescence of the OM fraction (Fig. 3). Thus the covalent modification protocol was nearly exclusive for FepA in the OM but allowed reactions with other proteins in both the periplasm and inner membrane.
FIGURE 4. FM labeling experiments.
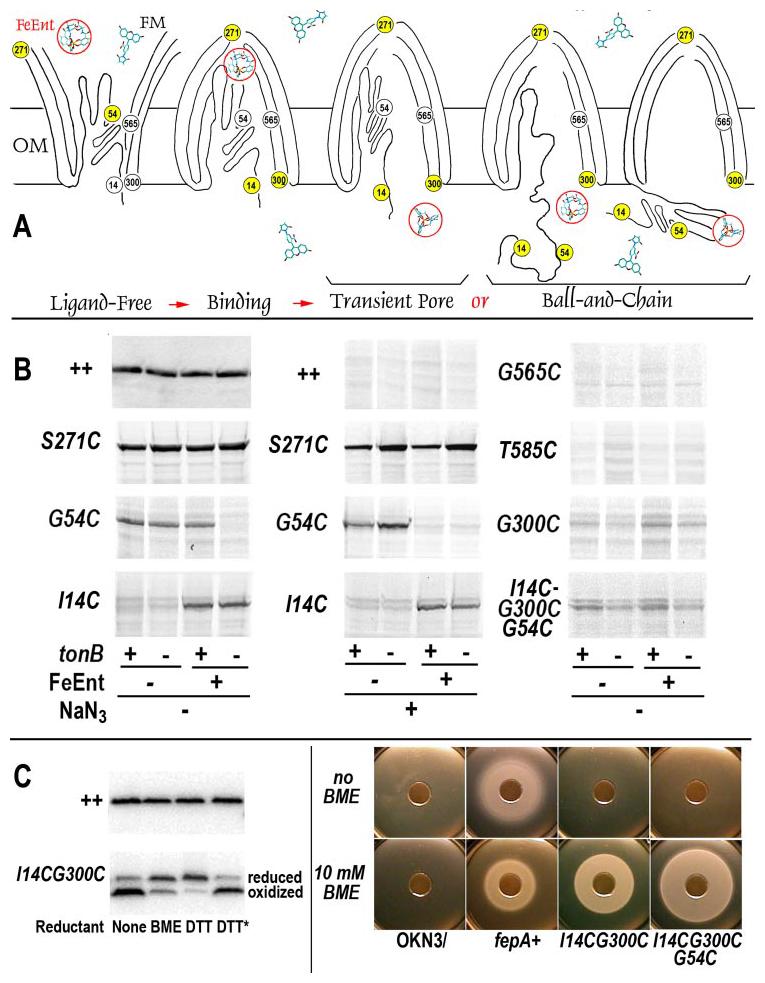
A, potential mechanisms. According to the Transient Pore postulate, fluors must enter FepA to react with the N-domain; in the ball-and-chain hypothesis, fluors may label the N-domain in the periplasm when either portions of (left), or the complete N-domain, dislodge from the channel. B, FM labeling of Cys substitution mutants. Wild-type FepA and its Cys mutant derivatives were exposed to FM in tonB+ (OKN3) and tonB (OKN13) bacteria, in the absence or presence of FeEnt (5 μm), and the absence or presence of NaN3 (10 mm). Cell lysates from the bacteria were resolved on SDS-PAGE gels, and the extent of fluorescence labeling was observed on the Storm Scan imager. Wild-type FepA (expression levels revealed by anti-FepA immunoblot, visualized with [125I]protein A, top left) was not labeled by FM (direct fluorescence scan, top center), whereas Cys substitution mutants (direct fluorescence scans) were modified. The reactivities of S271C, G54C and I14C were compared under various conditions, including the presence of NaN3 (10 mm). G54C was labeled during FeEnt uptake, but not when uptake was blocked by the absence of TonB or the presence of the poison (see text). These data were in contrast to G565C and T585C, which were not labeled, even though they reside on the interior surface of the β-barrel, close to residue 54. FepA and its mutants were consistently well expressed in the experiments (top left and data not shown). C, disulfide-bond formation between I14C and G300C. Genetically engineered Cys substitutions I14C and G300C formed a disulfide bond in vivo (left, visualized with anti-FepA antisera and [125I]protein A) that prevented transport of ferric enterobactin (panel B, center). If cells expressing I14CG300C, or the triple mutant I14C/G54C/G300C, were exposed to β-mercaptoethanol (5 mm) or dithiothreitol (10 mm), the disulfide was reduced, and the bacteria recovered their ability to transport FeEnt. The asterisk shows the results of treatment with oxidized dithiothreitol (10 mm).
FIGURE 5. Concentration dependence of FeEnt effects on FM labeling of G54C.
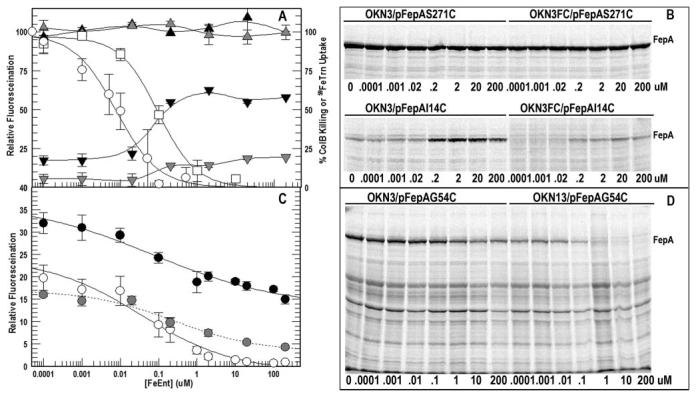
A, 5 × 108 cells of OKN3 (tonB+, ompF+, ompC+; black symbols) or OKN3FC (tonB+, ompF, ompC; gray symbols) harboring pFepAS271C (triangles) or pFepAI14C (inverted triangles) were exposed to FM (5 μm) in the presence of the noted concentrations of FeEnt for 15 min at 37 °C. The cells were collected by centrifugation, lysates of 108 bacteria were analyzed by SDS-PAGE, and the fluorescence of the FM-labeled FepA proteins was quantitated relative to the total labeling of all proteins in the cell lysate. The fluoresceination of surface residue S271C (mean of three trials) occurred at 26% of total cell labeling and was independent of host strain and [FeEnt]. The fluoresceination of periplasmic I14C (mean of three trials; expressed as percentage of S271C-FM) varied with host strain (presence or absence of OmpF and OmpC) and [FeEnt]. B, raw data from single experiments on the effect of [FeEnt] on FM labeling of S271C and I14C. Panel A also shows the concentration dependence of FeEnt inhibition of ColB killing (□) and 59FeTrn uptake (○), with fitted curves calculated by the IC50 algorithm of Grafit 5.013. C, 5 × 108 cells of OKN3 (black symbols), OKN3FC (gray symbols), and OKN13 (tonB, ompF+, ompC+; open symbols) carrying pFepAG54C were exposed to FM and analyzed for fluorescence as above. The labeling of FepAG54C was quantitated relative to total fluorescence in the lysate and normalized as a percentage of S271C-FM in the same host background. The figure shows the mean values for OKN3 (four trials), OKN3FC (three trials) and OKN13 (four trials) with fitted curves from the IC50 algorithm of Grafit 5.013. D, raw data from single experiments on the effect of [FeEnt] on FM labeling of FepAG54C in OKN3 and OKN13. In A and C, vertical bars on the data points represent the standard errors of the means.
Differential Accessibility of G54C during FeEnt Binding and Transport
From the initial data we selected six Cys substitutions in different locations for further study: I14C, G54C, S271C, G300C, G565C, and T585C (Fig. 4B). When exposed to FM under physiologically relevant conditions (for 15 min at 37 °C, with or without FeEnt, in tonB+ or tonB cells, in the absence or presence of NaN3), the FM-labeling patterns at these sites documented conformational changes that occur during FeEnt transport, as follows. (a) FM did not label wild-type FepA under any conditions, but it labeled S271C (in L3) in all conditions, whether FeEnt was present or absent, and whether the bacteria were energy deficient, sufficient, tonB, or tonB+. (b) In tonB+ or tonB cells, in the absence of FeEnt, FM reacted with G54C, located on the surface of the N-domain, approximately midway through the OM bilayer. The reactivity of this site in the absence of FeEnt may reflect the fact that FepA adopts an open conformation in vivo (25), allowing FM to encounter G54C from the exterior (see also below). (c) In tonB bacteria, in the presence of FeEnt, FM did not react with G54C. TonB-deficient cells bind but do not transport FeEnt, and binding of the ferric siderophore converts FepA to a closed conformation (25) with restricted access to the receptor's interior from either the exterior or the periplasm. (d) In energized, tonB+ cells, in the presence of FeEnt, G54C was labeled. These cells bound and transported FeEnt, and the labeling of G54C under transport conditions showed that during FeEnt uptake the N-domain surface regained its accessibility to FM. It was unlikely that FM reacted with G54C from the exterior in the presence of FeEnt (10 μm; ∼105-fold excess over Kd (0.2 nm)), because the ligand preferentially occupies the receptor and excludes the fluor (see also below). If so, then FM gained access to G54C from the periplasmic space when FepA underwent motion associated with transport. Consistent with this interpretation, inhibition of energy metabolism by NaN3 (panel B, center), and hence FeEnt uptake, eliminated the fluoresceination of G54C. These data suggested that FM penetrated the OM and labeled G54C from the periplasm. Although both models rationalize increased accessibility of G54C from the periplasm during transport, other experiments provided more insight. (e) In tonB+ or tonB cells, FM modified I14C, but more strongly in the presence of FeEnt. These data confirmed that FM reached the periplasm, where it was sufficiently concentrated to label I14C at the rim of the FepA β-barrel. It also weakly labeled A33C, G300C, and T666C (Figs. 1 and 2), all of which reside at the periplasmic interface. FeEnt binding and/or transport improved the susceptibility of both I14C and G300C, probably because ligand binding relocates the TonB-box away from the β-barrel wall, as it does in other LGP (15-17). (f) To test the possibility that FM entered the interior of FepA when a pathway to the periplasm formed during uptake, we monitored its reactivity with G565, S569C, and T585C on the interior surface of the β-barrel, in direct apposition to G54C. In all strains and circumstances, FM did not react with these three residues, showing that it does not gain access to G54C by entering the β-barrel. The labeling of G54C, but not G565C, S569C, or T585C, argued against the Transient Pore hypothesis, and supported the alternative, ball-and-chain model.
So, in various conditions and genetic backgrounds, FM differentially labeled Cys at positions 14 and 54. Residue G54C was susceptible when FepA was unoccupied, and the occupancy of its binding site by FeEnt (seen in tonB cells) protected G54C from modification. When the metal complex passed through FepA, G54C regained accessibility to FM, and the resulting labeling reaction had the same tonB and energy dependence as transport itself, suggesting that FM modified G54C when FepA underwent motion associated with transport. The fluoresceination of I14C showed the presence of FM in the periplasm, where it may gain access to G54C either when a channel opens to the receptor's interior or when the N-domain extrudes into the periplasm. However, the lack of FM reactivity with G565C, S569C, and T585C, close to G54C in the protein's interior, argued against the former explanation, which is the central postulate of the Transient Pore mechanism.
Effect of a Disulfide Bond between I14C and G300C
The formation of a disulfide bond between I14C and G300C blocked the transport of FeEnt (Fig. 4C), and reduction of the same disulfide restored the ability of FepA to transport the ligand. When we also introduced G54C into the I14C/G300C background, FepA was similarly dysfunctional in transport, and reduction with β-mercaptoethanol restored the FeEnt transport ability of I14C/G54C/G300C. FM labeled this mutant at a reduced level that was unaffected by the presence of FeEnt. Hence restraint of the N-domain inside the barrel decreased fluoresceination of G54C below the level that was seen during transport.
Concentration Dependence of FeEnt Inhibition of FM Labeling
To interpret the fluoresceination of G54C in terms of the FepA transport mechanism, we determined the pathway by which FM accessed the Cys sulfhydryl. Although thermodynamic arguments made it likely that FeEnt occupied and blocked the receptor vestibule, it was still conceivable that FM reacted with G54C from the exterior even during FeEnt transport, especially because the surface loops of the receptor undergo conformational changes during ligand internalization (25, 47, 50). For insight into these alternatives, we studied the concentration dependence of FeEnt-mediated inhibition of ligand uptake. First, the competitive inhibition of 59FeTrn uptake and colicin B killing (Fig. 5) by FeEnt verified the ability of the ferric siderophore to occupy the receptor binding site and exclude other molecules. Both FeTrn and colicin B themselves adsorb to FepA with high affinity (Kd = 10−8 m (28) and 10−7 m (26), respectively), but FeEnt effectively inhibited 59FeTrn uptake and ColB-mediated killing (Table 1; IC50 values of 8 nm and 90 nm, respectively (Fig. 5A)). So, even at nanomolar levels, FeEnt excluded the other molecules from binding and transport interactions with FepA, strongly suggesting that it blocks the access of FM to G54C from the exterior at equivalent or higher concentrations.
TABLE 1. Concentrations of FeEnt resulting in 50% inhibition of solute interaction with FepA (IC50).
Each experiment was performed three or four times, and the mean values were analyzed by the IC50 algorithm of Grafit 5.013 (Erithacus Ltd., Surrey, UK).
| Solute | Host strain | fepA allele | IC50 |
|---|---|---|---|
| μm | |||
| 59FeTrn | OKN3 | + | 0.008 ± 0.002 |
| Colicin B | OKN3 | + | 0.09 ± 0.02 |
| FM | OKN3 | G54C | 0.07 ± 0.07 |
| FM | OKN13 | G54C | 0.04 ± 0.02 |
| FM | OKN3FC | G54C | 0.25 ± 0.24 |
We next characterized the ability of FM to penetrate the OM and react in the periplasm, by comparing modification of FepAS271C (on the cell surface) and FepAI14C (in the periplasm) in porin-containing E. coli strain (OKN3: ompF+ and ompC+) and porin-deficient cells (OKN3FC: ompF and ompC). S271C consistently reacted with FM to an extent that represented 26% of the total cellular labeling, and this level was unaffected by FeEnt or porin deficiency (in OKN3: 25.7%; in OKN3FC: 25.9% (Fig. 5A)). Even high concentrations of the ferric siderophore (200 μm) failed to impair the fluoresceination of the S271C sulfhydryl, which resides in the extremity of L3. Conversely, both FeEnt and porin deficiency affected the labeling of I14C. As previously seen (Figs. 1 and 2), FeEnt binding and transport enhanced the modification of I14C. 10 nm FeEnt was the threshold for this effect: below this concentration FM modified I14C at a low level (∼18% of that observed for S271C); above it the modification increased 3-fold (to ∼60% of S271C, or 16% of total cellular labeling (Fig. 5A)). Secondly, whether or not FeEnt was present, reduction of diffusional solute uptake into the periplasm by elimination of the general porins OmpF and OmpC caused a 70% decrease in the labeling of I14C. These data showed that significant amounts of FM entered the periplasm by transit through OmpF/C, that the general porin pathway was the major route for entry of the extrinsic fluorophore into the cell, and that in the absence of OmpF and OmpC sufficient reagent still reached the periplasm by other routes to sustain a 30% residual level of fluoresceination.
Lastly, we characterized the effects of FeEnt on the labeling of FepAG54C in wild-type, TonB- and OmpF/C-deficient bacteria. In tonB cells (OKN13/pFepAG54C) low concentrations of FeEnt inhibited, and high concentrations abrogated fluoresceination of G54C. The threshold again occurred at 10 nm, 100 nm FeEnt caused a 50% reduction, and above 1 μm the inhibition of labeling was complete (Fig. 5B). These inhibition data concurred qualitatively and quantitatively with the competitive effects of FeEnt on ColB killing and 59FeTrn uptake (Table 1), reiterating that at concentrations exceeding 10 μm FeEnt occupies the receptor protein, excluding its reception of other molecules. In tonB+ cells, on the other hand, the effects of FeEnt were different: OKN3/pfepAG54C bound and transported the ferric siderophore. In the presence of FM, as [FeEnt] rose above 10 nm the fluoresceination of G54C declined (with a consistent IC50 (Table 1)), but at ∼1 μm the FM-labeling level stabilized and did not further decrease, despite higher concentrations of the ligand. Even 200 μm FeEnt, 106-fold above Km, did not eliminate this residual reaction of FM with G54C. Fluoresceination still occurred, at a level that was ∼50% of that observed without FeEnt. Thus in the presence of sufficient FeEnt to block fluoresceination from the exterior, half of the labeling of G54C persevered in transport active, tonB+ cells.
The results suggested two labeling processes for G54C: one from the outside that was susceptible to, and one from the periplasm that was resistant to inhibition by FeEnt binding or transport. Experiments with the porin-deficient host strain confirmed this inference. In the absence of FeEnt FM labeled OKN3/pFepAG54C at a level that was 32% of OKN3/pFepAS271C (Fig. 5, A and C); porin deficiency (in OKN3FC) caused a 30% reduction in labeling of G54C in these conditions, indicating that most of the fluoresceination came from the external pathway (see “Discussion”). In the presence of saturating FeEnt, on the other hand, porin deficiency caused a 60% reduction in labeling of G54C, indicating that most of the fluoresceination came from the internal pathway. So, at 200 μm FeEnt, the majority of the residual FM labeling of G54C came from the periplasm. In these conditions residue G54C behaved like I14C, the periplasmic control site. Thus in the presence of saturating FeEnt, G54C was primarily labeled from the periplasmic side of the OM bilayer.
Rescue Experiments: Complementation of Fepβ by FepN
The fluorescence data suggested expulsion of the N-domain from the C-domain during transport, which implies that, for ongoing transport to occur, the N terminus must re-assemble within the empty β-barrel. We tested this expectation by separately expressing the cloned N- and C-domains in OKN3. The co-expression of these partial genes in OKN3/pFepNpFepβ created significant physiological stress for the bacteria, initially decreasing their doubling time in LB broth to 10 h (relative to 35 min for OKN3/pITS23). However, after 8 h in LB broth (density of 2 × 108 cells/ml), the culture shifted to faster growth (doubling time = 75 min) until it logged out (Fig. 6). OKN3/pFepNpFepβ was resistant to colicins B and D during the first, slow growth phase, but sensitive to the toxins during the second, rapid phase (Table 2). The data showed the rescue of Fepβ, which was itself resistant to colicin B in all conditions, by the co-expression of FepN, which presumably entered within the empty channel, allowing recognition and reception of colicin B. We did not observe FeEnt transport by the hybrid, re-assembled protein.
FIGURE 6. Expression of FepA, FepN, and Fep β in OKN3.
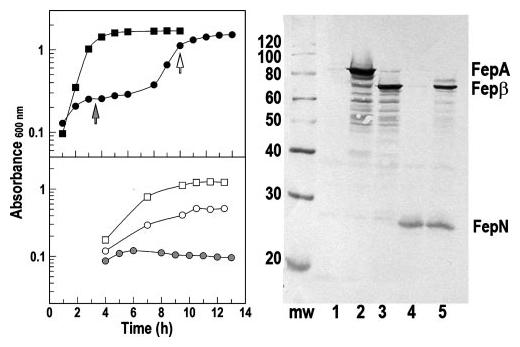
Left, growth in LB broth and MOPS minimal media. Top, OKN3 harboring pITS23 (squares) or pFepNpFepβ (circles) was inoculated into LB broth at 37 °C and shaken at 250 rpm. When expressing wild-type FepA, it grew without a lag and had a doubling time of 35 min; when separately synthesizing the N- and C-domains of FepA, OKN3 showed a first phase with doubling time of ∼10 h, and a second phase with doubling time of 75 min. Bottom, if subcultured from the first phase of LB growth into MOPS media (at the filled arrow), OKN3/pFepNpFepβ did not grow (filled circles), but when subcultured from the second phase (unfilled arrow), it had a doubling time of 3 h (open circles), only slightly slower than that of OKN3/pITS23 (2 h, open squares). Right, expression of FepA, FepN, and Fepβ (lane 5) in OKN3. After growth in LB or MOPS media for 6 h, lysates of OKN3 (lane 1) harboring pITS23 (lane 2), pFepβ (lane 3), pFepN (lane 4), and pFepN pFepβ (lane 5) were separated on SDS-PAGE gels and immunoblotted with anti-FepA MAbs (44) 45 and 41 (recognize epitopes in surface loop 4 (near residue 329 (27)) and the N-domain (residues 100–142 (48)), respectively. The immunoblot derives from bacteria grown in LB broth, but expression of FepA and its fragments was the same in all conditions, despite physiological differences in the two media at different stages of growth (see text).
TABLE 2. Co-expression of FepN and Fepβ.
E. coli strain OKN3, alone or harboring plasmids, was tested for FepA-dependent functions.
| Host strain/plasmidsa | Alleleb | ColBc | ColDc |
|---|---|---|---|
| OKN3 | fepA | R | R |
| OKN3/pITS23 | fepA+ | 106 | 105 |
| OKN3/pFepN | fepN-(1–151) | R | R |
| OKN3/pFepβ | fepβ-(1–17; 151–724) | R | R |
| OKN3/pFepNpFepβ | FepNfepβ | 4 × 104 | 3 × 103 |
| OKN13 | tonBfepA | R | R |
| OKN13/pITS23 | fepA+ | R | R |
| OKN13/pFepNpFepβ | fepNfepβ | R | R |
OKN3 contains an engineered deletion of the fepA structural gene (“Experimental Procedures”), pITS23 and pFepβ are derivatives of the low copy number vector pHSG575, and pFepN is a derivative of pUC18.
The fepN allele encodes residues 1–150 of wild-type FepA, under the control of its natural promoter; fepβ (fepAΔ18–150) encodes residues 1–17 followed by a deletion of residues 18–150 and continues with residues 151–724.
Purified colicin was diluted in a microtiter plate over a 108-fold range, and 5 μ l of each dilution was transferred to an LB agar plate, previously coated with 2.5 × 108 bacterial cells in LB top agar. The plates were incubated overnight at 37 °C. The tabulated values are the reciprocal of the final dilution that showed visible clearing of the bacterial lawn.
DISCUSSION
The differential labeling of engineered Cys sulfhydryl groups by FM reflected their chemical accessibility in vivo. These data defined distinct structural states in FepA during its transport reaction. Fluoresceination of some sites in FepA was energy-and TonB-dependent, illustrating the ability of the technique to characterize events that occur during transport. Furthermore, analysis of multiple sites in strains with altered permeability explained the labeling patterns: FM reacted with sites on the cell surface but also traversed general porin channels and modified residues in the periplasm. Its mass, 427 Da, is near the limit of general porin permeability but still consistent with transit through OmpF/C (41, 51). In an ompF, ompC strain, fluoresceination of the periplasmic residue I14C decreased 70%. This result verified the uptake pathway and demonstrated that FM reaches sufficient concentrations to modify sulfhydryls in the periplasm.
The differential modification of I14C reiterated the analytical utility of the labeling method. In the absence of FeEnt, interactions between the TonB-box and the β-barrel wall restricted access to I14C, but its susceptibility increased when FepA bound or transported FeEnt. These results fulfill the expectation of structural rearrangements in the TonB-box during FeEnt binding/transport. Similar motion occurs in FhuA (15, 16) and BtuB (17, 52) but was not seen in the FepA crystal structure (4) or by other methods. Thus in vivo, non-covalent bonds between the N-domain and the channel impair the reaction of FM with the I14C sulfhydryl, suggesting that they are strong enough to stabilize pore closure in the absence of ligand (53). When FeEnt binds, it releases this interaction, preparing the transporter for ligand internalization and coincidentally freeing I14C for reactivity with FM.
The unexpected reactivity of G54C led us to define the pathway by which FM encountered it. Experiments in a tonB strain, which does not transport ligands, were informative. In the absence of FeEnt, FM labeled G54C, and the presence of FeEnt blocked its modification, suggesting that the fluorophore accesses G54C from the exterior and that FeEnt in the binding site hinders this pathway. However, labeling of G54C during FeEnt transport implied access from the periplasmic route, and analysis of cell surface and periplasmic control residues in porin-containing and porin-deficient strains (especially the 70% reduction in modification of I14C in OKN3FC) confirmed this second pathway. In the absence of FeEnt, the OKN3FC background decreased modification of G54C by 30%, suggesting that in cells with a normal OM, 43% (30%/0.7) of labeling came from the periplasmic pathway and 57% from the exterior. But, in a transport-competent strain (OKN3; tonB+) saturating FeEnt inhibited 50% of G54C labeling, and porin deficiency eliminated 60% of the residual reactivity. Thus, during FeEnt transport 86% of the modification activity (60%/0.7) came from the periplasmic route: G54C was primarily labeled by periplasmic FM.
The activity of FM in the periplasm potentially discriminates between the Transient Pore and ball-and-chain mechanisms. Although it modified G54C during FeEnt transport, FM failed to label G565C, S569C, and T585C, a few angstroms away on the interior wall of the β-barrel. This result infers that the fluor does not enter the channel to react with G54C. These labeling data are consistent, nevertheless, with the alternative explanation: extrusion of the N-domain into the periplasm. The only potential caveat to this chain of evidence is the possibility that inaccessibility of G565C, S569C, and T585C arises from unforeseen conformational or steric factors within the β-barrel, which preclude their reaction with FM.
Besides the conclusions from accessibility of FepA to labeling, our experiments found additional grounds for the ball-and-chain mechanism. Elimination of transport and reduction in G54C labeling by disulfide bond formation between the N-domain, and the β-barrel is consistent with the ball-and-chain model. Comparable studies with FhuA reduced but did not block ferrichrome uptake, but the engineered disulfide bonds in that system formed less efficiently (54) than I14C/G300C in FepA. Finally, complementation of the empty channel by a separately expressed N terminus fulfills the model's expectation that the globular domain must repeatedly insert into the β-barrel. The functionality of the fragmented, self-assembled FepA protein, even at a fraction of wild-type levels, demonstrates the feasibility of this re-assembly reaction. The single-hit mechanism of colicin action (i.e. a single molecule may kill a bacterial cell) makes colicin killing the most sensitive measure of FepA function. In the conditions we employed, the E. coli OM contains ∼50,000 FepA proteins (28, 30, 38); thus the 4% level of activity from co-expression of the two domains indicates that ∼2000 copies of the N and C fragments successfully assembled in each cell. We did not see FeEnt uptake by the hybrid FepA protein, but this is not surprising because of the low level of re-assembly and probable low efficiency of transport by such a fragmented protein. Nevertheless, its ability to confer colicin sensitivity verified the functional reconstitution of the hybrid protein. Conversely, OKN3/pFepβ was colicin-resistant. Similar complementation was previously observed in the FhuA system (37), illustrating the general ability of LGP N-domains to insert into their β-barrels.
Despite the overall organization of FepA tertiary structure in ball-and-chain architecture, the potential removal of its globular domain from the channel during solute internalization faces an energetic barrier posed by potential hydrogen and ionic bonds between the N-domain and β-barrel. Two points are relevant here. The surface of the N-domain is hydrophilic, and the formation of new H-bonds, to other residues or to water in the periplasmic space, may compensate the energetic costs of breaking H-bonds to residues on the barrel wall. Secondly, a scaffold of ionic bonds in the protein interior may control N-domain motion in and out of the barrel. Charge attractions stabilize the N terminus within the pore, whereas charge repulsions (created by pH changes in the protein interior) may provide a force for its expulsion (53). Thus the N-domain may act as a door, hinged to the barrel and held in place by non-covalent bonds, until other forces supersede them and expel the globule, simultaneously transporting the ligand through the pore. Direct interactions with TonB, as seen in its co-crystallization with BtuB and FhuA (55, 56), represents a second potential trigger to initiate exit of the N-domain and ligand internalization.
Acknowledgments
We thank Ines Chen, David Dubnau, Helen Zgurskaya, Paul Cook, and Alain Charbit for critically reading the manuscript, Volkmar Braun for providing strain MB97, and Hiroshi Nikaido for providing strain HN705.
Footnotes
This work was supported in part by National Science Foundation Grant MCB-0414694 and National Institutes of Health Grant GM53836 (to P. E. K. and S. M. C. N.).
The abbreviations used are: OM, outer membrane; IM, inner membrane; FeEnt, ferric enterobactin; FM, fluorescein maleimide; MOPS, 4-morpholinepropanesulfonic acid; TBS, Tris-buffered saline; LGP, ligand-gated porin.
W. Rabsch, L. Ma, G. Wiley, F. Najar, B. Roe, W. Kaserer, B. Biel, M. Schmalley, S. M. Newton, and P. E. Klebba, manuscript submitted for publication.
REFERENCES
- 1.Pugsley AP, Reeves P. J. Bacteriol. 1976;126:1052–1062. doi: 10.1128/jb.126.3.1052-1062.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McIntosh MA, Earhart CF. Biochem. Biophys. Res. Commun. 1976;70:315–322. doi: 10.1016/0006-291x(76)91144-x. [DOI] [PubMed] [Google Scholar]
- 3.Wayne R, Frick K, Neilands JB. J. Bacteriol. 1976;126:7–12. doi: 10.1128/jb.126.1.7-12.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buchanan SK, Smith BS, Venkatramani L, Xia D, Esser L, Palnitkar M, Chakraborty R, van der Helm D, Deisenhofer J. Nat. Struct. Biol. 1999;6:56–63. doi: 10.1038/4931. [DOI] [PubMed] [Google Scholar]
- 5.Saier MH., Jr. J. Membr. Biol. 2000;175:165–180. doi: 10.1007/s00232001065. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong CM, Bezanilla F. J. Gen. Physiol. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nikaido H, Vaara M. Microbiol. Rev. 1985;49:1–32. doi: 10.1128/mr.49.1.1-32.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nikaido H. Microbiol. Mol. Biol. Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Masi DR, White JC, Schnaitman CA, Bradbeer C. J Bacteriol. 1973;115:506–513. doi: 10.1128/jb.115.2.506-513.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luckey M, Neilands JB. J. Bacteriol. 1976;127:1036–1037. doi: 10.1128/jb.127.2.1036-1037.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luckey M, Pollack JR, Wayne R, Ames BN, Neilands JB. J. Bacteriol. 1972;111:731–738. doi: 10.1128/jb.111.3.731-738.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bradbeer C, Woodrow ML, Khalifah LI. J. Bacteriol. 1976;125:1032–1039. doi: 10.1128/jb.125.3.1032-1039.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bradbeer C, Kenley JS, Di Masi DR, Leighton M. J. Biol. Chem. 1978;253:1347–1352. [PubMed] [Google Scholar]
- 14.Braun V, Wolff H. FEBS Lett. 1973;34:77–80. doi: 10.1016/0014-5793(73)80707-0. [DOI] [PubMed] [Google Scholar]
- 15.Locher KP, Rees B, Koebnik R, Mitschler A, Moulinier L, Rosenbusch JP, Moras D. Cell. 1998;95:771–778. doi: 10.1016/s0092-8674(00)81700-6. [DOI] [PubMed] [Google Scholar]
- 16.Ferguson AD, Hofmann E, Coulton JW, Diederichs K, Welte W. Science. 1998;282:2215–2220. doi: 10.1126/science.282.5397.2215. [DOI] [PubMed] [Google Scholar]
- 17.Chimento DP, Mohanty AK, Kadner RJ, Wiener MC. Nat. Struct. Biol. 2003;10:394–401. doi: 10.1038/nsb914. [DOI] [PubMed] [Google Scholar]
- 18.Wang CC, Newton A. J Bacteriol. 1969;98:1142–1150. doi: 10.1128/jb.98.3.1142-1150.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pugsley AP, Reeves P. J Bacteriol. 1977;130:26–36. doi: 10.1128/jb.130.1.26-36.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bradbeer C. J Bacteriol. 1993;175:3146–3150. doi: 10.1128/jb.175.10.3146-3150.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang CC, Newton A. J. Biol. Chem. 1971;246:2147–2151. [PubMed] [Google Scholar]
- 22.Guterman SK, Dann L. J. Bacteriol. 1973;114:1225–1230. doi: 10.1128/jb.114.3.1225-1230.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reynolds PR, Mottur GP, Bradbeer C. J. Biol. Chem. 1980;255:4313–4319. [PubMed] [Google Scholar]
- 24.Postle K. J. Bioenerg. Biomembr. 1993;25:591–601. doi: 10.1007/BF00770246. [DOI] [PubMed] [Google Scholar]
- 25.Scott DC, Newton SM, Klebba PE. J. Bacteriol. 2002;184:4906–4911. doi: 10.1128/JB.184.17.4906-4911.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Payne MA, Igo JD, Cao Z, Foster SB, Newton SM, Klebba PE. J. Biol. Chem. 1997;272:21950–21955. doi: 10.1074/jbc.272.35.21950. [DOI] [PubMed] [Google Scholar]
- 27.Cao Z, Qi Z, Sprencel C, Newton SM, Klebba PE. Mol. Microbiol. 2000;37:1306–1317. doi: 10.1046/j.1365-2958.2000.02093.x. [DOI] [PubMed] [Google Scholar]
- 28.Annamalai R, Jin B, Cao Z, Newton SM, Klebba PE. J. Bacteriol. 2004;186:3578–3589. doi: 10.1128/JB.186.11.3578-3589.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newton SM, Allen JS, Cao Z, Qi Z, Jiang X, Sprencel C, Igo JD, Foster SB, Payne MA, Klebba PE. Proc. Natl. Acad. Sci. U. S. A. 1997;94:4560–4565. doi: 10.1073/pnas.94.9.4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newton SM, Igo JD, Scott DC, Klebba PE. Mol. Microbiol. 1999;32:1153–1165. doi: 10.1046/j.1365-2958.1999.01424.x. [DOI] [PubMed] [Google Scholar]
- 31.Merianos HJ, Cadieux N, Lin CH, Kadner RJ, Cafiso DS. Nat. Struct. Biol. 2000;7:205–209. doi: 10.1038/73309. [DOI] [PubMed] [Google Scholar]
- 32.Usher KC, Ozkan E, Gardner KH, Deisenhofer J. Proc. Natl. Acad. Sci. U. S. A. 2001;98:10676–10681. doi: 10.1073/pnas.181353398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1972. [Google Scholar]
- 34.Sprencel C, Cao Z, Qi Z, Scott DC, Montague MA, Ivanoff N, Xu J, Raymond KM, Newton SM, Klebba PE. J. Bacteriol. 2000;182:5359–5364. doi: 10.1128/jb.182.19.5359-5364.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klebba PE, McIntosh MA, Neilands JB. J. Bacteriol. 1982;149:880–888. doi: 10.1128/jb.149.3.880-888.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Datsenko KA, Wanner BL. Proc. Natl. Acad. Sci. U. S. A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Braun M, Endriss F, Killmann H, Braun V. J. Bacteriol. 2003;185:5508–5518. doi: 10.1128/JB.185.18.5508-5518.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott DC, Cao Z, Qi Z, Bauler M, Igo JD, Newton SM, Klebba PE. J. Biol. Chem. 2001;276:13025–13033. doi: 10.1074/jbc.M011282200. [DOI] [PubMed] [Google Scholar]
- 39.Hashimoto-Gotoh T, Franklin FC, Nordheim A, Timmis KN. Gene (Amst.) 1981;16:227–235. doi: 10.1016/0378-1119(81)90079-2. [DOI] [PubMed] [Google Scholar]
- 40.Smit J, Kamio Y, Nikaido H. J. Bacteriol. 1975;124:942–958. doi: 10.1128/jb.124.2.942-958.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nikaido H, Rosenberg EY. J. Bacteriol. 1983;153:241–252. doi: 10.1128/jb.153.1.241-252.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rutz JM, Abdullah T, Singh SP, Kalve VI, Klebba PE. J. Bacteriol. 1991;173:5964–5974. doi: 10.1128/jb.173.19.5964-5974.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ames GF. J. Biol. Chem. 1974;249:634–644. [PubMed] [Google Scholar]
- 44.Murphy CK, Kalve VI, Klebba PE. J. Bacteriol. 1990;172:2736–2746. doi: 10.1128/jb.172.5.2736-2746.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vakharia HL, Postle K. J. Bacteriol. 2002;184:5508–5512. doi: 10.1128/JB.184.19.5508-5512.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Deleted in proof.
- 47.Cao Z, Warfel P, Newton SM, Klebba PE. J. Biol. Chem. 2003;278:1022–1028. doi: 10.1074/jbc.M210360200. [DOI] [PubMed] [Google Scholar]
- 48.Rutz JM, Liu J, Lyons JA, Goranson J, Armstrong SK, McIntosh MA, Feix JB, Klebba PE. Science. 1992;258:471–475. doi: 10.1126/science.1411544. [DOI] [PubMed] [Google Scholar]
- 49.Sugawara E, Nikaido H. J. Biol. Chem. 1992;267:2507–2511. [PubMed] [Google Scholar]
- 50.Jiang X, Payne MA, Cao Z, Foster SB, Feix JB, Newton SM, Klebba PE. Science. 1997;276:1261–1264. doi: 10.1126/science.276.5316.1261. [DOI] [PubMed] [Google Scholar]
- 51.Nikaido H, Rosenberg EY, Foulds J. J. Bacteriol. 1983;153:232–240. doi: 10.1128/jb.153.1.232-240.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cadieux N, Phan PG, Cafiso DS, Kadner RJ. Proc. Natl. Acad. Sci. U. S. A. 2003;100:10688–10693. doi: 10.1073/pnas.1932538100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klebba PE. Front. Biosci. 2003;8:1422–1436. doi: 10.2741/1233. [DOI] [PubMed] [Google Scholar]
- 54.Eisenhauer HA, Shames S, Pawelek PD, Coulton JW. J. Biol. Chem. 2005;280:30574–30580. doi: 10.1074/jbc.M506708200. [DOI] [PubMed] [Google Scholar]
- 55.Shultis DD, Purdy MD, Banchs CN, Wiener MC. Science. 2006;312:1396–1399. doi: 10.1126/science.1127694. [DOI] [PubMed] [Google Scholar]
- 56.Pawelek PD, Croteau N, Ng-Thow-Hing C, Khursigara CM, Moiseeva N, Allaire M, Coulton JW. Science. 2006;312:1399–1402. doi: 10.1126/science.1128057. [DOI] [PubMed] [Google Scholar]
- 57.Cobessi D, Celia H, Folschweiller N, Schalk IJ, Abdallah MA, Pattus F. J. Mol. Biol. 2005;347:121–134. doi: 10.1016/j.jmb.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 58.Neidhardt FC, Bloch PL, Smith DF. J. Bacteriol. 1974;119:736–747. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]