

Abstract
The promyelocytic leukemia protein PML is known to form nuclear multiprotein complexes which are compromised in several pathogenic conditions including acute promyelocytic leukemia. We show that in cells infected with a single stranded RNA virus, which relocates PML bodies to the cytoplasm, the infected cells are more resistant to serum starvation induced apoptosis than their uninfected counterparts. Antisense PML oligonucleotides increase cell survival under serum deprivation conditions indicating that PML is directly involved in the apoptotic activity. Transient transfection studies have indicated that this pro-apoptotic activity of PML is mediated through the zinc binding region known as the RING finger. Viral attack of PML nuclear bodies appears to allow the virus to deregulate host cell apoptotic machinery in order to establish chronic infection.
1. Introduction
The promyelocytic leukemia protein (PML) was first identified as a fusion protein with the retinoic acid receptor alpha (RARα) in acute promyelocytic leukemia (APL) patients [1–3]. The PML protein forms nuclear multiprotein complexes with at least seven other proteins [4–12]. The nuclear bodies are disrupted in the APL disease state [10–12]. Further, the protein has been shown to have growth and transformation suppressor activity [13]. PML contains a set of conserved cysteine-rich zinc binding regions known as the RING, Bl and B2 B-boxes. These domains are followed closely by a leucine coiled coil region forming the tripartite or RBCC motif [14,15]. Mutagenesis studies have shown that these domains are necessary for nuclear body formation [16,17]. The loss of nuclear body formation has been linked to a loss of growth and transformation activity of PML [13]. The molecular function of these bodies is yet to be established.
PML nuclear bodies are compromised in several kinds of viral infections [19–24]. We have shown that the majority of PML bodies are relocated to the cytoplasm in NIH 3T3 and HeLa cells when infected with a single stranded RNA virus, lymphocytic choriomeningitis virus (LCMV) [18]. This virus is known to establish chronic infection in tissue culture cell lines [25]. Many viruses subvert growth suppression and apoptotic mechanisms of host cells during infection [26]. Obviously controlling these cellular processes is crucial for viruses during establishment of chronic infection. Viruses can reset the cell cycle as part of this process [26,27]. In this report we have monitored the effect of LCMV infection on the cell cycle by observation of changes in distribution of the cell cycle marker, proliferating cell nuclear antigen (PCNA). These studies indicate that the subcellular distribution of PCNA changes after infection.
Previous work has shown that apoptosis and the cell cycle are intimately linked [27]. Cell cycle checkpoints may act as a switch between proliferation and apoptosis by sensing deregulation of the cell cycle [27]. Cell lines stably over-expressing the PML-RARα fusion protein, where most of the fusion protein is located in the cytoplasm, have a reduced commitment to apoptosis when compared to normal cells [28]. Furthermore, PML is found in the cytoplasm of hepatocarcinoma cells [29]. The deletion of the nuclear localization signal in PML is found to not only cause relocation of PML to the cytoplasm but also is correlated with a loss of its growth and transformation suppression activities [13]. Thus PML appears to be found in the cytoplasm in cells which are not responding to normal growth control. As PML is redistributed in LCMV infection, we investigated its role in serum deprivation induced apoptosis. In this report we show that PML has a pro-apoptotic function mediated through its RING domain. Viral redistribution of PML nuclear bodies may allow the virus to deregulate host cell apoptotic machinery.
2. Materials and methods
2.1. Infection of cells with LCMV
Infections were carried out as described [18]. Briefly, NIH 3T3 cells were infected with 1 plaque forming unit of LCMV Armstrong per cell. Infection was verified by staining with anti-LCMV antibody as described [18] (data not shown). Cells were grown on coverslips and fixed in acetone before staining. Immunofluorescence studies were carried out as described [17]. A monoclonal antibody to PCNA (Santa Cruz) was used at a dilution of 1:100 with a FITC (fluorescent isothioconjugate) goat anti-mouse secondary (Vector). The images were collected on a Leica confocal laser microscope.
2.2. Serum withdrawal of infected cells
NIH 3T3 cells were grown in 10% FBS/DMEM. Cells were infected as above in 24 well dishes. Cells were washed in serum free media and then fresh serum free media was added. Cells were washed every two days until harvested for counting. Parallel experiments were carried out in uninfected cells. Live cells were counted using trypan blue exclusion to differentiate between live and dead cells. Four fields of view were counted per well. Variation is no greater than ± 5% for each point.
2.3. The effect of antisense PML on survival of serum starved NIH 3T3 cells
Oligonucleotides were added to a total concentration of 3 μM or the equivalent volume of serum free media was added for control wells. Cells were grown in 12 well dishes in 10% FBS/DMEM, pretreated for 1 h with the appropriate oligonucleotide or with media only. Cells were then rinsed with serum free media and fresh serum free media was added. At this stage the oligonucleotides were reapplied in case of loss during the washing step. After 18 h, cells were harvested, and counted. Trypan blue exclusion was used to differentiate live from dead cells. Experiments were carried out in quadruplicate and four fields were counted for each so that each number is a result of at least 16 measurements. Error bars indicate standard deviations. Oligonucleotides were obtained from Genosys. The antisense sequence was as follows: 5′ C(S)GGAGATCGGGCGGGTGCAGG(S)C 3′. (S) indicates the presence of a phosphorothioate bond. The sense sequence was the corresponding sequence for the alternate strand. As an additional control, the effects of an unrelated oligonucleotide were monitored as well. This oligonucleotide was to a portion of a viral protein which is absent from uninfected cells. The sequence was as follows: 5′ GGCGGCTGCCATGGCCATGGGTCAAGGCAAGTCC 3′.
Whole cell lysates were obtained from cells as treated above using standard procedures [32]. Total protein concentration was determined in order that equivalent amounts of each lysate were used. Cell lysates were boiled in reducing loading buffer and subjected to 20% SDSPAGE. The gels were blotted onto immobilin P (Millipore) and probed with a PML polyclonal antibody which has been characterized extensively elsewhere [14,16]. Bound antibody was visualized using the ECL method (Amersham).
2.4. Over-expression of PML decreases survival of serum starved cells
NIH 3T3 cells were transfected using lipofectamine (Gibco) according to the manufacturer’s instructions. One μg of plasmid DNA was used for each transfection. Cells were grown in 12 well plates. Transfections were carried out in quadruplicate. After treatment with lipofectamine, cells were allowed to recover overnight in 10% FBS/DMEM. This was followed by washing the cells in serum free media. Cells were washed every second day until they were harvested for counting on either day 3 or day 6. Cells were counted using trypan blue exclusion to establish they were live. At least four fields from each well were counted. Thus, each experimental point is the result of at least 16 measurements. Error bars indicate the standard deviations. Cell counts from transfection with vector with no insert and cells treated with lipofectamine but no DNA gave identical results within statistical error. Therefore the effect of the lipofectamine has been taken into account in these measurements. The constructs used in these assays are as described in [16,18]. Briefly, the mutations in PML were for the site 1 RING mutation, Cys9Cys12ΔAla [16] and the coilless mutation was made in the pml mammalian expression construct by deletion of the BssHII fragment as described [18]. This deleted both the leucine coiled coil and a portion of the B2 B-box. The double point mutation in Z site 2 of the RING domain was made as described [18]. The mutation was Cys32Cys35ΔPheGly. Aside from abrogating the metal binding ability of the RING, upon analysis of the other RING structures, the introduction of the Phe should be structurally destabilizing. The PML RING had been shown to be structured only in the presence of zinc [16]. Mutation in the zinc binding ligands therefore destroys the structure of the domain. All constructs were sequenced to confirm no spurious mutations occurred during processing.
2.5. Death by apoptosis during serum starvation was confirmed using standard methods
It is well established that NIH 3T3 cells undergo apoptosis under serum deprivation conditions [30,31]. Cells were grown on coverslips, transfected and underwent serum withdrawal as described above. These coverslips were stained with YOYO (Molecular Probes), a fluorescent dye which binds to nucleic acid (as described by the manufacturer). Condensation of DNA is a characteristic of apoptotic as opposed to necrotic death [30]. Fluorescence was monitored using confocal laser microscopy.
3. Results and discussion
3.1. The subcellular distribution of PCNA is altered by infection
In order to investigate how widespread the effects of LCMV infection are on the cell, the subcellular distribution of the cell cycle marker, proliferating cell nuclear antigen (PCNA), was monitored using immunofluorescence and confocal laser microscopy. In Fig. 1 cells are shown which have been stained with PCNA in normal NIH 3T3 cells (A) and those infected with LCMV (B). In panel A, the staining is nuclear punctate. In B, there is a cytoplasmic punctate pattern with significantly less nuclear staining. There appears to be a correlation between LCMV infection and redistribution of PCNA.
Fig. 1.
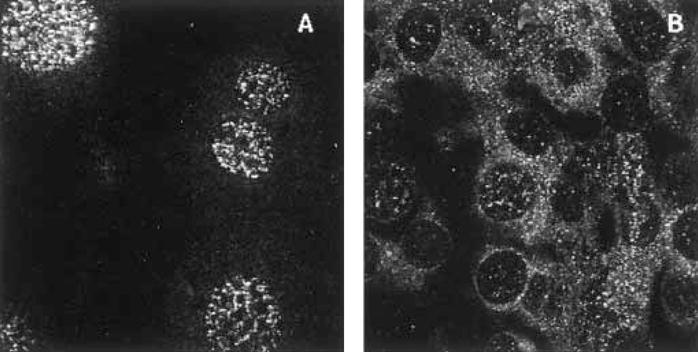
The effect of LCMV infection on the subcellular distribution of the cell cycle marker PCNA. Panels A and B show uninfected and infected NIH 3T3 cells respectively. Cells were infected with 1 plaque forming unit per cell of LCMV Armstrong for 100 h as described previously [18]. The images were collected on a Leica con-focal laser microscope. Magnification was 100X.
3.2. Infection delays cell death during serum starvation
Our previous work showed that PML was relocated to the cytoplasm during LCMV infection. Displacement of PML to the cytoplasm has been associated with loss of normal growth control [13,18,28,29], therefore we investigated whether viral infection had increased the cells resistance to apoptosis induced by serum starvation. NIH 3T3 cells are known to undergo apoptotic death during serum starvation [30,31]. Results in Fig. 2 indicate that infected cells survived longer than their uninfected counterparts suggesting that the relocation of PML may be important to survival of both the host and the virus. Infected cells were present until day 10 whereas no uninfected cells remained by day 6.
Fig. 2.
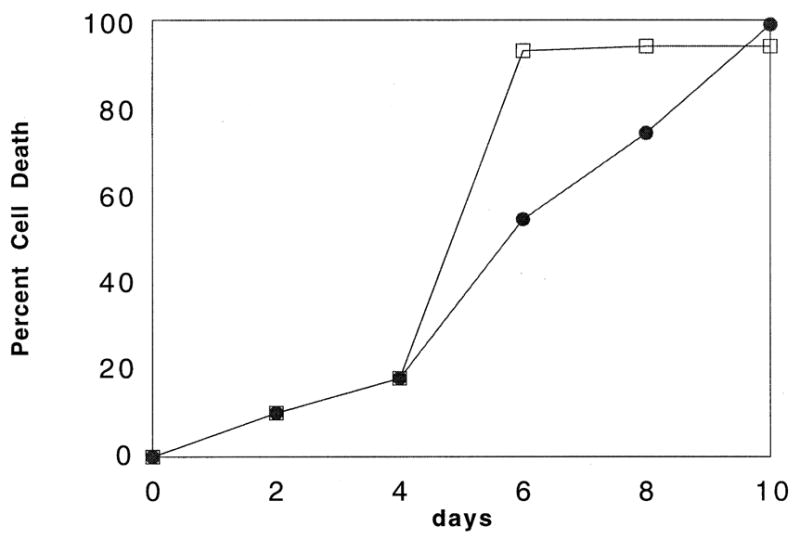
LCMV infection increases survival in serum starved NIH 3T3 cells. Cells were infected as in Section 2. Live cells were counted using trypan blue exclusion. Each data point represents the average trypan blue cell count of three wells in a 24 well plate. Four fields of view were counted per well. Variation is no greater than ± 5% for each point. Data from infected cells are indicated by filled circles and normal cells by open squares.
3.3. Treatment with PML antisense and sense oligonucleotides
To attribute this effect to the PML protein we carried out antisense experiments. NIH 3T3 cells were treated with either media, antisense oligonucleotide to PML or the corresponding sense oligonucleotide. Cells were then serum starved, harvested and counted (Fig. 3). Cells treated with the antisense oligonucleotide survived longer than cells treated with the sense oligonucleotide, an unrelated oligonucleotide or media only. The difference was statistically significant with 71 ± 8% cell survival in antisense treated cells while cells treated with sense, unrelated oligonucleotide or media only the average value is 53 ± 7%. There seems to be no difference in the survival of cells treated with the sense oligonucleotide, the unrelated oligonucleotide or cells that were only serum withdrawn. Cell death was occurring by apoptosis as confirmed using standard methods (see Section 2 and below).
Fig. 3.
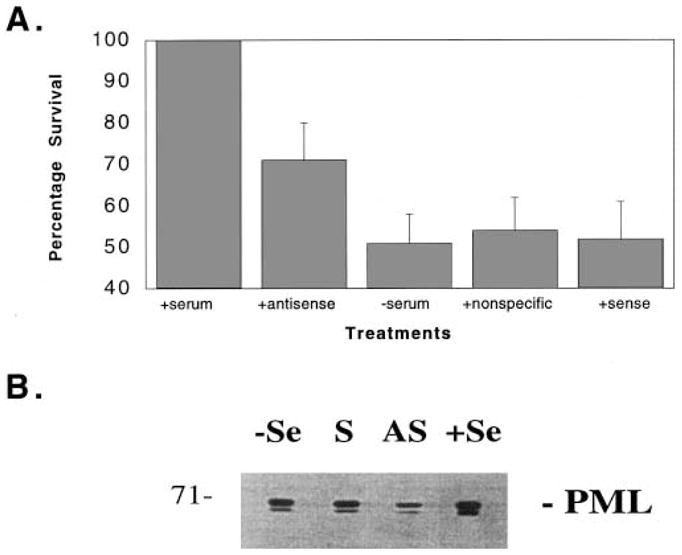
A: The effect of antisense PML on survival of serum starved NIH 3T3 cells. Oligonucleotides were added to a total concentration of 3 μM. Cells were grown in 12 well plates in 10% FBS/DMEM, pre-treated for 1 h with the appropriate oligonucleotide or with media only. Cells were then rinsed with serum free media and new serum free media was added. After 18 h, cells were harvested, and counted. Experiments were carried out in quadruplicate and four fields were counted for each. Error bars indicate standard deviations. B: Protein expression levels of PML were monitored using standard techniques in cells as treated in A. Total cell lysates were used where the total protein concentration was normalised prior to loading onto gels. Standard Western blots were obtained and probed with a PML polyclonal antibody (see Section 2). –Se indicates serum withdrawn, +Se, with serum, S, serum withdrawn cells treated with sense oligonucleotide and AS with antisense.
Cells treated identically to those used in the above cell counts were assayed for PML protein levels. Cells were lysed and protein concentrations normalised to ensure that equal amounts of total protein were loaded onto gels. Protein levels were monitored by Western blot analysis. Fig. 3B shows that levels of PML were downregulated in serum withdrawn cells treated with the antisense oligonucleotide relative to those treated with sense oligonucleotide, media or control cells which were not serum withdrawn. The lower band is the phosphorylated form of PML.
3.4. Mutational analysis and transfection studies indicate PML is pro-apoptotic and this activity is mediated through its RING domain
The effect of serum starvation was monitored in transient transfection studies carried out with PML. PML mutants which have a nuclear diffuse distribution were also investigated. These include a double point mutation in the RING finger (RING), which decreases its ability to fold [16,17] and a deletion of the coil and B2 B-box (CL) [13,16,17]. Similar mutations mitigated the growth suppression activity of PML [13]. Furthermore, the effect of the viral protein Z was studied as this protein is sufficient to translocate PML to the cytoplasm during LCMV infection [18]. Z is a 90 residue protein which contains a RING domain and directly binds PML [18]. A double point mutation in the RING of the Z protein was also studied. The Z protein is required for genome synthesis and viral replication of LCMV [25]. A summary of mutations is given in Fig. 4A.
Fig. 4.
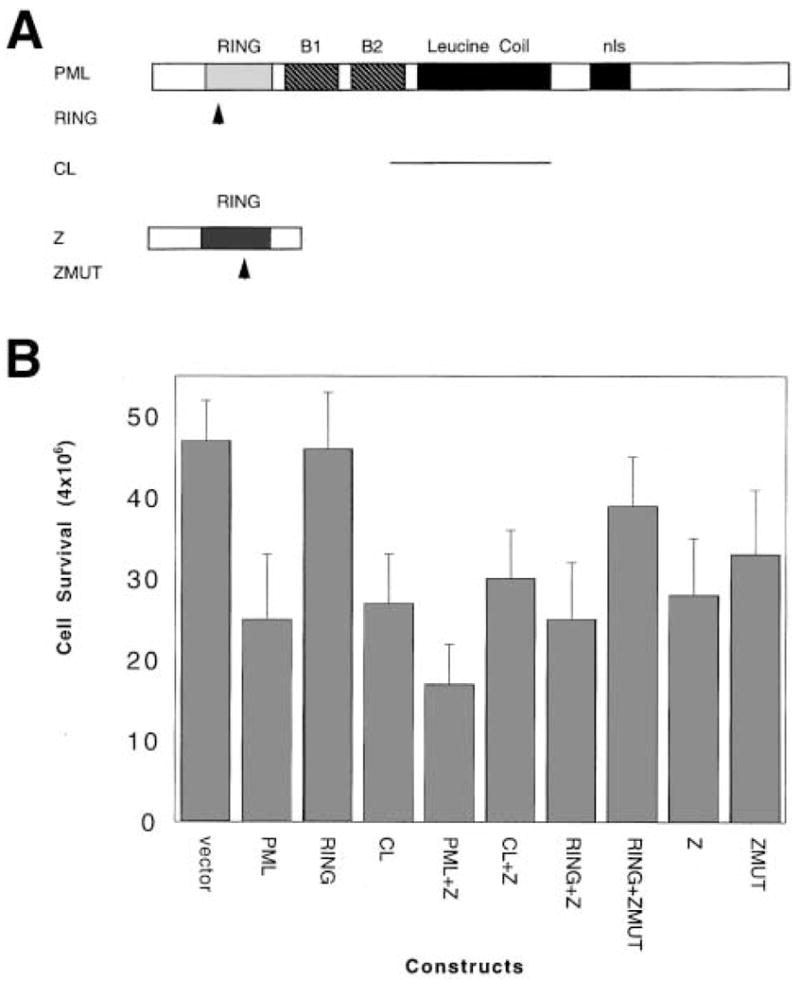
Over-expression of PML decreases survival of serum starved cells. Cells were transfected with the constructs described in Section 2. A summary of constructs is shown in A. Arrows indicate the position of point mutations and lines represent deletions. Results of cell counts are shown in B. Transfected cells were allowed to recover overnight before serum withdrawal. For full details of mutations see text. Each experimental point is the result of at least 16 measurements. The data shown are from cells serum starved for three days before counting. Error bars indicate the standard deviations.
The results of these experiments are given in Fig. 4B. Transfection of vector alone indicates the baseline level for cell survival. Transfection with PML resulted in a nearly two-fold decrease in cell survival versus vector alone. A similar result is observed for the CL mutant. The most striking result is that the PML RING mutant has the same cell survival as vector alone. Previous work indicates that these mutants express similarly to wild-type protein [16–18] so differences cannot be attributed to significantly lower levels of the RING mutant in the cells. Addition of the viral protein alone decreases cell survival but this decrease is not significantly augmented by co-transfection with PML or the CL mutant. Interestingly, co-transfection with the RING mutant and Z also decreases survival. Co-transfection of the RING with the mutant Z gives an effect intermediate between RING alone and RING with Z. Mutation in the Z-RING leads to increased cell survival relative to Z.
It was confirmed that cells were undergoing apoptotic and not necrotic death in transfection studies with PML and RING. In all cases we observed the characteristic shrinkage of cells and nuclear condensation as monitored by staining cells with the nucleic acid dye YOYO (see Section 2). Fig. 5A shows cells which have not been serum withdrawn stained with YOYO. Intense nuclear staining is observed. In panel B, cells were mock transfected and subsequently serum withdrawn as was carried out for the experiments described in Fig. 4. The white arrows indicate condensed nucleic acid signals characteristic of apoptotic nuclei [30]. In panels C and D cells have been transfected with either the RING mutant or PML (C and D respectively) prior to serum withdrawal. Apoptotic nuclei are observed in both cases but are more numerous in the cells transfected with PML, consistent with the results shown in Fig. 4. Qualitatively, the relative percentages of apoptotic nuclei observed was consistent with that observed in Fig. 4. Cells transfected with RING had less apoptotic nuclei and more live cells than those transfected with PML.
Fig. 5.
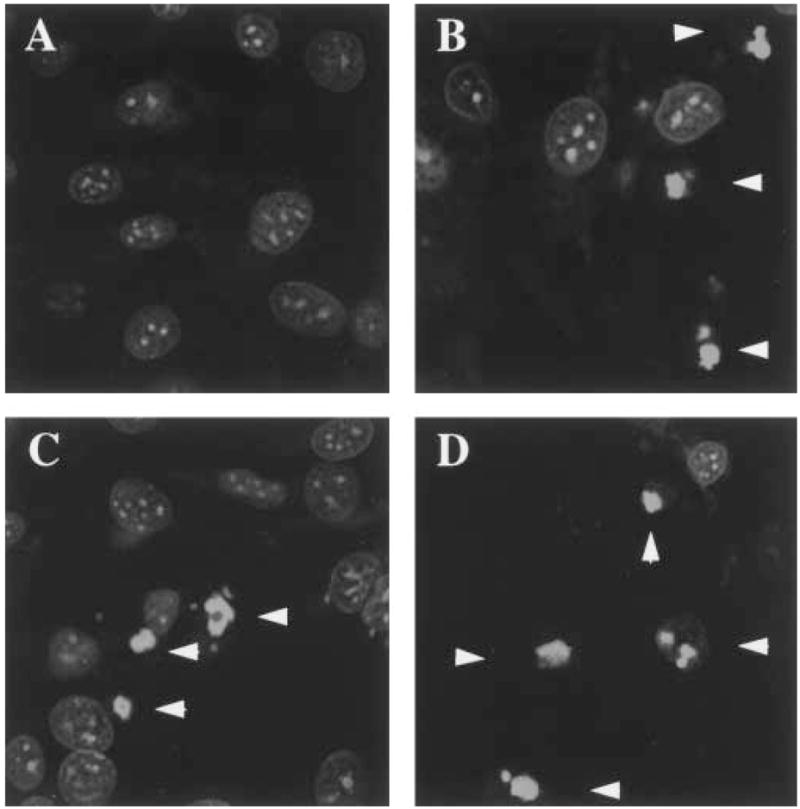
NIH 3T3 cells apoptose under serum deprivation conditions. Cells were stained with YOYO, a nucleic acid binding dye, and images are from a confocal laser microscope. White arrows indicate apoptotic bodies. Cells were treated identically to those in Fig. 4. Cells were treated as follows: plus serum control (A), serum withdrawn cells mock transfected (B), serum withdrawn cells transfected with the RING mutant (C) and serum withdrawn cells transfected with PML. The objective was 40 × with zoom of 3.282 (A), 3.282 (B), 3.282 (C) and 3.313 (D).
3.5. Summary
We show that unlike growth and transformation suppression, the apoptotic activity of PML is not linked to the formation of nuclear bodies. Both PML which forms nuclear bodies and the CL mutant which has a nuclear diffuse phenotype decreased cell survival significantly. Alternately, the PML RING mutant which also has a diffuse nuclear distribution does not decrease cell survival under these conditions. We have previously shown that co-transfection of CL and Z led to a nuclear and cytoplasmic punctate pattern [18]. The Z protein appears to have a dominant effect over mutations in PML in terms of its subcellular distribution which result in the reformation of PML bodies but these bodies are present in both the nucleus and cytoplasm where this virus replicates. Further our previous studies indicate that the interaction between PML and Z is direct and that Z binds the first 45 residues in PML which are directly adjacent to the PML RING [18]. Therefore it is possible that the Z protein and PML could be bound to each other and a third component could be shared between their respective RING domains.
Our mutational studies indicate that some component that PML RING normally binds to, dissociates as a result of the mutation in the RING. This component when not bound to the PML protein either increases cell survival or inhibits the apoptotic pathways in play during serum starvation. The data suggest that Z binds to the same component as its RING. Although Z decreases cell survival, LCMV infection increases it under these conditions. The acquisition of this RING binding component may be necessary for viral persistence. The presence of the other viral proteins during infection may prevent Z from killing the cells and permit the virus to establish chronic infection. For instance, Z is known to interact directly with the LCMV nucleocapsid protein [25].
The apoptotic activity of PML appears to be mediated through its RING domain. The RING family of proteins now consists of over 80 members [14,15]. Several proto-oncogene proteins have RING domains including the breast cancer gene product BRCA1. The tripartite subfamily, with the RING, B-boxes and leucine coiled coil domain also comprise of several oncogenes including transforming factor 18 and the ret finger protein [14,15]. It appears that the PML RING domain is targeted by many viruses in order to commandeer host cell components important in deregulation of apoptosis. Our data suggest that although growth, transformation suppression and apoptosis are logically linked processes in the cell, their mechanisms with regard to PML are slightly different. It is sufficient to disrupt nuclear body formation in order to lose growth and transformation suppression activity [13] but the RING domain itself must be effected to alter the apoptotic activity of PML.
Acknowledgments
We are indebted to Graeme Carlile for helpful discussions. We thank K. Howe, E. Solomon and P. Freemont for the kind gift of the PML polyclonal antibody. K.L.B.B. acknowledges financial support from MRC (Canada) MT-13608 and M.S.S. N.I.H. RO1 AI32107.
References
- 1.Goddard AD, Borrow J, Freemont PS, Solomon E. Science. 1991;254:1371–1373. doi: 10.1126/science.1720570. [DOI] [PubMed] [Google Scholar]
- 2.Pandolfi PP, et al. Oncogene. 1991;6:1285–1292. [PubMed] [Google Scholar]
- 3.Kakizuka A, et al. Cell. 1991;66:663–674. doi: 10.1016/0092-8674(91)90112-c. [DOI] [PubMed] [Google Scholar]
- 4.Boddy MN, et al. Oncogene. 1996;13:971–982. [PubMed] [Google Scholar]
- 5.Ascoli CA, Maul GG. J Cell Biol. 1991;112:785–795. doi: 10.1083/jcb.112.5.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stuurman N, et al. J Cell Sci. 1992;101:773–784. doi: 10.1242/jcs.101.4.773. [DOI] [PubMed] [Google Scholar]
- 7.Maul GG, Ishov AM, Everett RD. Virology. 1996;217:67–75. doi: 10.1006/viro.1996.0094. [DOI] [PubMed] [Google Scholar]
- 8.Korioth F, Gieffers C, Maul GG, Frey J. J Cell Biol. 1995;130:1–13. doi: 10.1083/jcb.130.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desbois C, Rousset R, Bantignies F, Jalinot P. Science. 1996;273:951–953. doi: 10.1126/science.273.5277.951. [DOI] [PubMed] [Google Scholar]
- 10.Koken MH, et al. EMBO J. 1994;13:1073–1083. doi: 10.1002/j.1460-2075.1994.tb06356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dyck JA, et al. Cell. 1994;76:333–343. doi: 10.1016/0092-8674(94)90340-9. [DOI] [PubMed] [Google Scholar]
- 12.Weis K, et al. Cell. 1994;76:345–356. doi: 10.1016/0092-8674(94)90341-7. [DOI] [PubMed] [Google Scholar]
- 13.Le XF, Yang P, Chang KS. J Biol Chem. 1996;271:130–135. doi: 10.1074/jbc.271.1.130. [DOI] [PubMed] [Google Scholar]
- 14.Borden KLB, Freemont PS. Curr Opin Struct Biol. 1996;6:395–401. doi: 10.1016/s0959-440x(96)80060-1. [DOI] [PubMed] [Google Scholar]
- 15.Saurin AJ, Borden KLB, Boddy MN, Freemont PS. Trends Biochem Sci. 1996;246:208–213. [PubMed] [Google Scholar]
- 16.Borden KLB, et al. EMBO J. 1995;14:1532–1541. doi: 10.1002/j.1460-2075.1995.tb07139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borden KLB, et al. Proc Natl Acad Sci USA. 1996;93:1601–1606. [Google Scholar]
- 18.Borden KLB, CampbellDwyer EJ, Salvato MS. submitted. [Google Scholar]
- 19.Maul GG, Everett RD. J Gen Virol. 1994;75:1223–1233. doi: 10.1099/0022-1317-75-6-1223. [DOI] [PubMed] [Google Scholar]
- 20.Kelly C, Van Driel R, Wilkinson GWG. J Gen Virol. 1995;76:2887–2893. doi: 10.1099/0022-1317-76-11-2887. [DOI] [PubMed] [Google Scholar]
- 21.Szekely L, et al. J Virol. 1996;70:2562–2568. doi: 10.1128/jvi.70.4.2562-2568.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishov AM, Maul GG. J Cell Biol. 1996;134:815–826. doi: 10.1083/jcb.134.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doucas V, et al. Genes Dev. 1996;10:196–207. doi: 10.1101/gad.10.2.196. [DOI] [PubMed] [Google Scholar]
- 24.Puvion-Dutilleul F, et al. Exp Cell Res. 1995;218:9–16. doi: 10.1006/excr.1995.1125. [DOI] [PubMed] [Google Scholar]
- 25.Salvato MS, Rai SK. Arenaviruses, Topley and Wilson's Medical Microbiology and Microbial Infections. Chapter 31. Arnold Publishers; London, UK: 1996. [Google Scholar]
- 26.Thompson C. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 27.King KL, Cidlowski JA. J Cell Biochem. 1995;58:175. doi: 10.1002/jcb.240580206. [DOI] [PubMed] [Google Scholar]
- 28.Roagia D, et al. Leukemia. 1995;9:1467–1472. [PubMed] [Google Scholar]
- 29.Terris B, et al. Cancer Res. 1995;55:1590–1597. [PubMed] [Google Scholar]
- 30.Jacobson MD, et al. Nature. 1993;361:365. doi: 10.1038/361365a0. [DOI] [PubMed] [Google Scholar]
- 31.Raff MC. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- 32.Harlow E, Lane D. Antibodies. A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1988. [Google Scholar]