

Abstract
Superoxide-mediated clastogenesis is characteristic for various chronic inflammatory diseases with autoimmune reactions and probably plays a role in radiation-induced clastogenesis and in the congenital breakage syndromes. It is consistently prevented by exogenous superoxide dismutase (SOD), but not by heat-inactivated SOD, indicating that the anticlastogenic effect is related to the catalytic function of the enzyme. Increased superoxide production by activated monocytes/macrophages is followed by release of more long-lived metabolites, so-called clastogenic factors, which contain lipid peroxidation products, unusual nucleotides of inosine, and cytokines such as tumor necrosis factor α. Since these components are not only clastogenic, but can stimulate further superoxide production by monocytes and neutrophils, the genotoxic effects are self-sustaining. It is shown here that anticlastogenic effects of exogenous SOD are preserved despite extensive washing of the cells and removal of all extracellular SOD. Using flow cytometry and confocal laser microscopy, rapid adherence of the fluorescently labeled enzyme to the cell surface could be observed with slow uptake into the cell during the following hours. The degree of labeling was concentration and time dependent. It was most important for monocytes, compared with lymphocytes, neutrophils, and fibroblasts. The cytochrome c assay showed significantly diminished O2− production by monocytes, pretreated with SOD and washed thereafter. The preferential and rapid binding of SOD to monocytes may be of importance not only for the superoxide-mediated genotoxic effects, described above, but also from a therapeutic standpoint. It can explain the observation that beneficial effects of injected SOD lasted for weeks and months despite rapid clearance of the enzyme from the blood stream according to pharmacodynamic studies.
Keywords: chromosomal breakage, clastogenic factors, monocytes, fluorescein isothiocyanate–superoxide dismutase
Anticlastogenic (i.e., chromosomal) breakage preventing properties of superoxide dismutase (SOD) were reported soon after the discovery of the enzyme. Swedish authors observed that the enzyme, alone or in combination with catalase, decreased the frequency of radiation-induced or spontaneously occurring chromosome breaks in human lymphocyte cultures (1, 2). Later, Brawn and Fridovich (3) reported that strand breakage in supercoiled Escherichia coli DNA, exposed to a xanthine–xanthine oxidase system, was prevented partially with SOD alone and completely with a combination of SOD and catalase. They concluded that O2− and H2O2 collaborated in the production of OH· to cause strand breakage in DNA in a Haber–Weiss-type reaction. In recent years, the role of Fenton-type reactions was proposed to explain hydroxyl–radical mediated DNA damage by interaction of peroxides with iron-binding sites on DNA (4). Studies done on cellular systems showed that these genotoxic effects were preventable by iron chelators and the hydroxyl scavenger dimethyl sulfoxide, but not by SOD (5, 6). In contrast to this, our laboratory drew attention to indirect action mechanisms, in which O2− appeared to play a primary role, since the damage was regularly prevented by SOD alone, while catalase was not or irregularly protective (7, 8). Since inactivated SOD was not protective, the anticlastogenic effect should be related to the catalytic function of the enzyme.
Chromosomal breakage and rearrangement in cell cultures from patients with chronic inflammatory diseases with autoimmune reactions, such as progressive systemic sclerosis, rheumatoid arthritis, systemic lupus erythematosus, Crohn disease, ulcerative colitis, and disseminated sclerosis was prevented by SOD, indicating that superoxide radicals were involved in the clastogenic process (9). Also, the chromosomal instability and the increase in sister chromatid exchanges in cell cultures from Bloom syndrome patients could be reduced to control values by addition of SOD (150 units/ml) to the medium (10). The strongest argument for the implication of superoxide in these clastogenic effects was the observation that exposure of blood cultures to a xanthine oxidase reaction, photoreduction of flavins or a phorbol 12-myristate 13-acetate (PMA)-stimulated respiratory burst resulted in chromosomal breakage and sister chromatid exchanges.This could be consistently prevented by addition of SOD (30–150 units/ml), while catalase was irregularly protective (11–13).
Because it did not seem likely that extracellularly produced O2− would reach the nucleus without being scavenged by the intracellular SOD abundantly available in the cytosol, we proposed the formation of secondary clastogenic substances as an explanation. The reports of radiation biologists (14, 15) that the chromosome damage observed in irradiated persons is accompanied by transferable clastogenic plasma factors prompted us to look for such clastogenic factors (CF) in the plasma of patients with the above-mentioned diseases accompanied by “spontaneous” chromosome damage. These studies showed that CF are indeed regularly present in the plasma and in supernatants of patients’ cell cultures (8, 10, 16). Transferable clastogenic materials of low molecular weight were also isolated from supernatants of the above mentioned in vitro systems, in which chromosome damage was produced by generation of superoxide from various sources. Formation of CF was regularly prevented, when the cells were cultivated in presence of SOD. The clastogenic effects of preformed CF, after transfer into other cell cultures, were also prevented by SOD, indicating that superoxide was involved not only in the formation of these clastogenic plasma components, but also in their clastogenic action (17, 18). It could also be deduced from these studies that CF are cellular products. Indeed, exposure of cell-free culture media or serum did not result in clastogenic ultrafiltrates. Biochemical analysis of ultrafiltrates of plasma or culture supernatants identified various chromosome-damaging substances, including lipid peroxidation products (17–19), inosine triphosphate (20), and cytokines such as tumor necrosis factor α (8, 16), while hydrogen peroxide was not detectable (8, 19).
It could be shown with the cytochrome c assay that certain clastogenic components have also superoxide stimulating properties (21). Given that superoxide generation leads to the formation of clastogenic substances generating themselves superoxide, the system is self-sustaining and therefore responsible for long-lasting genotoxic processes, which may lead to cancer and autoimmune disease (7, 8). In this model of an auto-sustaining O2− generating process, SOD protects by interrupting the vicious circle. The present work shows that exogenous SOD exerts its protective effects not only in the extracelluler space, but that cells remain protected after careful removal of all extracellular SOD.
MATERIALS AND METHODS
Cells.
Venous blood from healthy blood donors was anticoagulated with heparin. Polymorphonuclear neutrophils (PMNs) and mononuclear cells were separated by dextran sedimentation and Ficoll/Hypaque density gradient centrifugation. Pure monocytes were prepared by adherence on plastic dishes. Human skin fibroblasts from semiconfluent cultures were detached from the Falcon plastics with a cell scraper. Single cell suspensions were obtained by successive passages through needles with decreasing diameter.
Cell Culture.
For cytogenetic studies, 1 × 106 mononuclear cells, obtained by density gradient centrifugation, were incubated in 5 ml tissue culture medium 199 (Sanofi, Paris), supplemented with 1 ml fetal calf serum from GIBCO. Cell division was promoted by the addition of 1 μg/ml phytohemagglutinin (Wellcome Diagnostics). After 70 h of incubation, mitoses were arrested in metaphase by addition of colchicine for another 2 h. Slides were prepared for chromosomal analysis according to classical cytogenetic proceduces and stained with Giemsa. The metaphases were examined under the microscope for the presence of gaps, breaks, fragments, exchange figures, and other types of aberrations.The frequency of aberrations was compared between sham-treated and clastogen-treated cultures (22).
The clastogen chosen for these experiments was inosine triphosphate (ITP), previously shown to be a component of CF and to act via superoxide-mediated mechanisms, since SOD, but not inactivated SOD, was regularly anticlastogenic (20). ITP (from Sigma) was dissolved in distilled water to a final concentration of 20 μM. For anticlastogenesis, Cu-Zn SOD from bovine erythrocytes (Palleau Production, Chateau-Landon, France) was added to the medium to a final concentration of 150 cytochrome c units/ml.The SOD activity in cell supernatants and culture media before and after washing of the cells was measured spectrophotometrically by monitoring the inhibition of cytochrome c reduction at 550 nm (23).
Preparation of Fluorescently Labeled SOD.
Fluorescein isothiocyanate (FITC; Sigma) was covalently conjugated to the SOD (Palleau Production) by constant stirring in 50 mM Na2C03 at 4°C for 1 h. The FITC–SOD was purified by Sephadex G-25 chromatography with 10 mM potassium phosphate buffer (pH 7.4) as the eluent.
Flow Cytometry.
Cells (1.5 × 106) were incubated in PBS at 37°C in presence of FITC–SOD at various concentrations and during various exposure times, as indicated for each experiment in Results. To avoid nonspecific binding, the cells were pretreated with 5% fetal calf serum during 20 min before exposure to FITC–SOD. After incubation, cells were washed three times in PBS to eliminate all residual free SOD.
FITC goat anti-mouse immunoglobulin fragments [F(ab′)2] were used as a negative control. Cytochalasin B (Sigma) was used at a 200 μM concentration.
Analysis was performed on a FACScan (Becton Dickinson). In the mononuclear cell suspension, monocytes and lymphocytes were gated according to forward scatter and side scatter. Large cells were monocytes (>99% CD14+), small cells were lymphocytes (>90% CD3+ or CD19+). PMN were distinguished from contaminating red blood cells as large granular cells according to forward and side scatter. FITC fluorescence was detected at 520 nm. Data were collected and analyzed by Lysis II software (Becton Dickinson).
High Resolution Confocal Scanning Laser Microscopy of Monocytes.
The mononuclear cell fraction, containing monocytes plus lymphocytes, was incubated during various times (15 min to 3 h) with FITC–SOD (150 units/ml), as described above for flow cytometry. After three washes, the cells were brought to adherence on plastic dishes. After 1 h of adherence, the monocyte monolayer was rinsed with PBS to remove the lymphocytes. Fixation of monocytes was realized with 4% formol for 30 min. Nuclei were stained with chromomycin a3 (Sigma) for 30 min at room temperature in the dark. After rinsing with PBS, the monocyte layer was mounted in Fluoprep (Bio-Mérieux, Marcy l’Etoile, France). Confocal fluorescent images were obtained with a MRC-600 confocal scanning laser microscope (Bio-Rad), equipped with a 25-mW multiline Argon ion laser (Ion Laser Technology, Salt Lake City, UT), a z-stepping motor, a 80386/87 MS/DOS Nimbus microcomputer (Research Machines, Oxford, U.K.) and som (Bio-Rad) software. The Optiphot (Nikon, Tokyo) was equipped with a ×60 plan apochromat (Nikon) objective with a 1.4 numerical aperture. Immersion oil with a 1.515 refraction index was used. The (x,y) scanning was usually made with a 1.0 electronic zoom. To reduce “noise,” a Kalman filter recording an average of 10 images/sec was used for confocal imaging. Excitation at 488 nm and emission at LP 515 nm were the wavelengths for detecting the localization of the FITC–SOD. For imaging of the chromomycin A3-stained DNA of nuclei, a combination of two filters was used: one filter for excitation at 458 nm and a high-pass from 550 nm for emission.
Measurement of Superoxide Production by PMN and Monocytes.
The cells were incubated in 1 ml of 80 μM ferricytochrome c solution in PBS during 60 min and stimulated with PMA (Chemicals for Cancer Research, Eden Prairie, MN; final concentration 30 ng/ml), after pretreatment with SOD at various concentrations during 2 h and 3 washes. The absorbance of the supernatants was measured in a spectrophotometer at 550 nm, and the concentration of reduced cytochrome c was determined by using an extinction coefficient of 21,000 M−1·cm−1. The total O2− production was calculated, assuming that the reduction reaction is stoichiometric (24). Additional dishes were prepared containing 150 units/ml of S0D over the entire incubation period to determine the SOD-inhibitable fraction of cytochrome c reduced. Reaction mixtures without cells served as blanks.
RESULTS
Inhibition of Clastogenesis in SOD-Pretreated Cells Resuspended in SOD-Free Medium.
In five independent experiments, human lymphocyte cultures were exposed to ITP at a final concentration of 20 μM. This concentration induced 22.2 ± 3.85 structural chromosomal aberrations per 100 cells, similar to our previous study (20). The aberration rates were significantly (P < 0.01) reduced in the cell cultures which had been set up with cells preincubated with SOD (150 units/ml medium) and resuspended in fresh medium without SOD for exposure to the clastogen (Table 1). The measurements of SOD activity in the cell supernatants showed a reduction from 150 units/ml to 12.0 ± 2.73 units/ml after the first wash. SOD activity was no longer detectable after the second wash, which had reduced the concentration again 10 times.
Table 1.
Inhibition of ITP-induced clastogenic effects by pretreatment with SOD
| Exp. | Chromosomal aberrations/100 cells
|
||
|---|---|---|---|
| − SOD | + SOD | Control | |
| 1 | 20 | 10 | 10 |
| 2 | 29 | 16 | 4 |
| 3 | 16 | 8 | 8 |
| 4 | 24 | 10 | 6 |
| 5 | 22 | 5 | 10 |
| Mean | 22.2 ± 3.9 | 9.8 ± 3.2 | 7.5 ± 2.1 |
Experimental conditions: concentration, 150 units/ml; incubation time, 1 h; three washes; resuspension in fresh medium; absence of extracellular SOD in culture medium.
Flow Cytometry Shows Cellularly Bound FITC–SOD.
At a constant incubation time of 1 h, the fluorescent labeling increased with the SOD concentration. Monocytes regularly showed more labeling than lymphocytes and PMN, for which only the highest concentration of 1500 units/ml yielded significant results (Fig. 1). The degree of labeling was not only concentration-dependent, but also time-dependent (Fig. 2). No fluorescence labeling was observed on erythrocytes in four independent experiments after incubation times of 1 or 2 h with SOD concentrations of 150 units/ml (data not shown). The contamination with erythrocytes therefore did not influence the results for PMN. Preliminary experiments had shown that lysis of erythrocytes with 0.83% NH4Cl reduced the binding of FITC–SOD to PMN. Labeling of lymphocytes (SOD concentration, 150 units/ml) was not improved for activated cells, after stimulation with phytohemagglutinin (1 μg/ml) during 1, 4, and 24 h prior to incubation with FITC–SOD.
Figure 1.
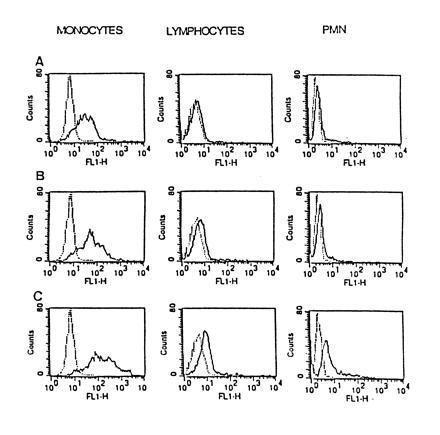
Flow cytometry. At a constant incubation time of 1 h, the fluorescent labeling of the FITC–SOD-treated cells increased with enzyme concentration (A, 150 units/ml; B, 750 units/ml; C, 1500 units/ml). Monocytes regularly showed more labeling than lymphocytes and PMN, for which only the highest SOD concentration yielded significant results.
Figure 2.
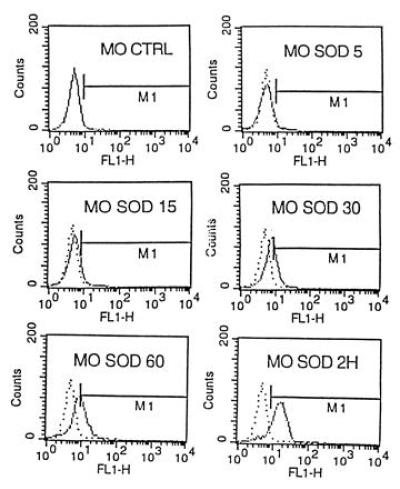
Flow cytometry. The fluorescent labeling of monocytes increased with exposure time at a constant concentration of 150 units/ml FITC–SOD.
Fibroblasts showed significant FITC fluorescence in three independent experiments (Fig. 3). Use of trypsine for detachment of the cells prevented the fluorescence labeling of the fibroblasts. Preincubation of cells with cytochalasin B during 4 h diminished the fluorescence but did not completely suppress it (Fig. 4).
Figure 3.
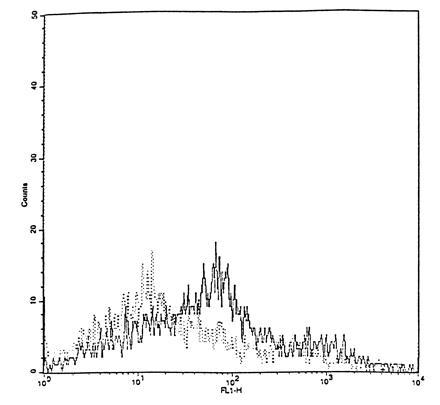
Flow cytometry. Not only blood cells, but also human skin fibroblasts are fluorescently labeled after exposure to FITC–SOD (concentration, 150 units/ml; incubation time, 2 h).
Figure 4.
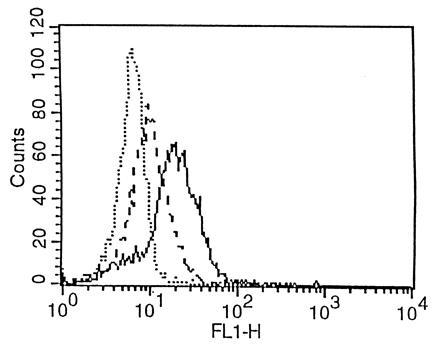
Flow cytometry. Cytochalasin B, an inhibitor of endocytosis, diminishes FITC-related fluorescence, but does not suppress it (intermediate labeling pattern, ---).
All experiments were performed at 37°C, since preliminary results had shown absence of fluorescence when the cells were incubated with FITC–SOD at 4°C. No fluorescence labeling of monocytes was observed with FITC-labeled goat anti-mouse immunoglobulin fragments (negative control).
Confocal Laser Microcopy Shows Fluorescence on the Membrane and in the Cytoplasm.
FITC–SOD-labeled monocytes showed a specific pattern of fluorescence, which was absent in controls. The intensity of fluorescence was time-dependent. For monocytes incubated with FITC–SOD for 15 min up to 1 h, the fluorescence was limited to the membrane. After a 2- to 3-h incubation period, FITC fluorescence could also be seen in the cytoplasm (Fig. 5), as ascertained upon focusing through the cell. No fluorescence was detectable at the nuclear level.
Figure 5.
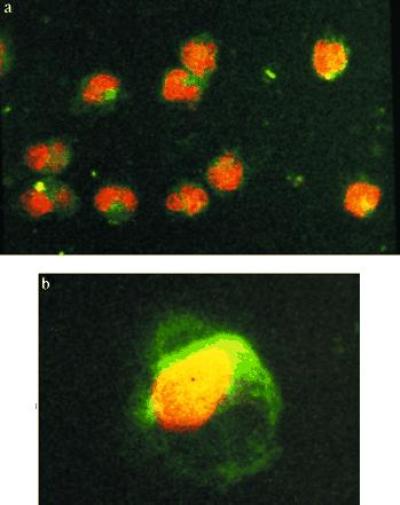
Confocal laser microscopy. After incubation during 15 min in presence of FITC–SOD (concentration, 150 units/ml), the surface of the monocytes show green fluorescence (a). After 3 h, FITC fluorescence could also be seen in the cytoplasms upon focusing through the cell. In b, a 1-μm-thick focal section taken in the middle of the cell. No labeling of the nucleus is seen.
PMA-Stimulated Superoxide Production Is Reduced After Pretreatment with SOD, but More for Monocytes Than for PMN.
In seven independent experiments, the PMA-stimulated superoxide production by PMN and monocytes from a same donor was compared for SOD-pretreated cells and nonpretreated cells (Table 2). Without preincubation, PMN (2.5 × 106) produced a mean of 55.6 ± 5.5 nmol of superoxide during 60 min upon stimulation with PMA. Addition of free SOD (150 units/ml) regularly reduced the values to those of blanks, indicating that the cytochrome c reduction was due entirely to superoxide. In comparison with the unprotected cells, the superoxide production by SOD-pretreated PMN, resuspended in SOD-free PBS, was reduced, but the differences did not reach statistical significance. In contrast to PMN, the inhibition of superoxide production by SOD-pretreated monocytes was more important. At the highest concentration of 1500 units/ml, the average reduction was 63.2% (range 53–85%), compared with 11% for PMN.
Table 2.
PMA-stimulated superoxide production by monocytes and PMN
| SOD | Monocytes*
|
Neutrophils†
|
|||||
|---|---|---|---|---|---|---|---|
| 0‡ | 300 | 1500 | 0 | 150 | 750 | 1500 | |
| I | 11.42 | 4.85 | 55.78 | 59.45 | |||
| II | 28.41 | 16.37 | 8.94 | 55.02 | 46.36 | 40.98 | |
| III | 31.51 | 18.75 | 14.85 | 48.26 | 43.88 | 46.30 | 44.12 |
| IV | 31.37 | 24.79 | 49.50 | 48.03 | 43.36 | 45.46 | |
| V | 28.37 | 9.90 | 4.20 | 54.17 | 50.74 | 49.79 | 50.12 |
| VI | 40.00 | 30.51 | 18.80 | 64.12 | 61.26 | 59.12 | 59.97 |
| VII | 32.13 | 23.27 | 12.42 | 62.21 | 58.54 | 60.69 | 56.17 |
| Mean | 29.03 | 20.61 | 10.68 | 55.58 | 52.61 | 51.80 | 49.47 |
| SD | 8.68 | 7.18 | 5.75 | 5.47 | 7.03 | 7.77 | 7.38 |
The figures represent nmoles O2− produced during 1 h.
Data for 0.3–0.5 × 105 cells.
Data for 2.5 × 106 cells.
Data in this row across are in units/ml.
DISCUSSION
According to the cytogenetic results of the present study, SOD given as a pretreatment, followed by suspension of the carefully washed cells in fresh medium, provides as good protection against clastogenesis as the same quantity of enzyme present in the culture medium over the entire cultivation period. The observed anticlastogenic effects were certainly not due to incomplete removal of the SOD from the culture medium. Not only was enzyme activity not detectable biochemically in the culture medium, but also the calculated SOD concentrations after the successive washes were too low for anticlastogenic effects (25).The small (statistically not significant) difference between chromosomal aberration rates in SOD-pretreated cells compared with controls is probably due to genotoxic effects not related to superoxide production. In addition to its action as a primer of superoxide production by competent cells, ITP is known to be an inhibitor of DNA topoisomerases (26).
FITC labeling of the SOD showed that the protective effect was related to binding of the enzyme to the cells. Despite the washes, all cell types present in blood cultures (lymphocytes, monocytes, neutrophils), except erythrocytes, had become fluorescent, as demonstrated by flow cytometry. This was also observed for fibroblasts, a cell type currently used in studies of clastogenesis and anticlastogenesis. The degree of labeling was concentration- and time-dependent. It was most important for monocytes at similar concentrations and exposure times. The question, whether the enzyme was located only on the cell surface or whether it also entered the cell, was answered by confocal microscopy, demonstrating progressive penetration of the SOD into the cytoplasma of monocytes.
Intracellular uptake of SOD after binding of the enzyme to cell surfaces had been reported by others using radioactive or colloidal gold labeling of the enzyme, combined with electron microscopy (27–29). Only Beckman et al. (30) used FITC, as we did, and observed a punctuate distribution pattern in endothelial cells similar to that shown in Fig. 5 for monocytes. Liposome-encapsulated (27, 31) or chemically modified SODs such as acylated SOD (28) or PEG–SOD (30) were considered to penetrate better than the native enzyme. However, the poor uptake of native SOD by monkey kidney cells, hamster fibroblasts and Ehrlich ascites cells observed by others (27) may have been the consequence of trypsinization, shown to reduce considerably the binding of native FITC–SOD in our experiments. In addition, binding of SOD to cell surfaces may be better in single cell suspensions, as prepared for our study, than for fibroblasts incubated with SOD as a confluent cell layer. Erythrocytes also showed poor labeling with native SOD in other studies (27, 28), while SOD incapsulated in liposomes penetrated via a mechanism described as “invagination” (27). Receptor-mediated endocytosis was proposed as the mechanism for penetration of this large molecule by others on the basis of results with various inhibitors of endocytosis (29, 32). In the present study, cytochalasin B pretreatment, despite higher concentrations and longer exposure times than those used by these authors, diminished the fluorescence labeling, but did not completely suppress it. The binding sites appeared to be specific for SOD, as far as SOD binding was not inhibited by bovine serum albumin pretreatment of hepatocytes (29). Specificity of SOD binding sites is also suggested by our results. Despite pretreatment of the cells with fetal calf serum, a procedure known to inhibit the binding of FITC-labeled proteins, the cells showed FITC fluorescence. Absence of fluorescence on monocytes, exposed to FITC-labeled goat anti-mouse immunoglobulins, is in favor of this interpretation, too. The importance of membrane integrity for the binding process was documented by its inhibition after previous contact of the cells with trypsine or NH4Cl. The observation that FITC labeling of the cells was obtained only at 37°C can be interpreted in favor of a metabolically active process.
We could not find any data in the literature concerning uptake of SOD by monocytes. Since a role of monocytes in clastogenesis was evident in our previous work, the preferential binding of the enzyme to monocytes, in comparison to other cell types, was of particular interest to us. Monocytes from polyarthritis patients induced clastogenic effects in lymphocyte cultures from healthy persons (31). This was not observed when heterologous monocytes from healthy persons were added. However, when increasing numbers of monocytes from healthy donors were added to cultures from other healthy donors in presence of PMA, the clastogenic effects increased correspondingly (33). SOD prevented clastogenesis. Small amounts of superoxide elicit monocytes/macrophages to release tumor necrosis factor, which in turn stimulates these cells to produce more tumor necrosis factor (34). This mediator is one of the clastogenic components of CF and probably responsible in part for the vicious circle of CF formation (8). In addition, immunologically activated monocytes, as well as PMA-stimulated monocytes, release free arachidonic acid and various metabolites of the arachidonic acid cascade, of which certain have clastogenic properties and were found in CF preparations from culture supernantants (19, 35). In the present study we show that PMA-stimulated superoxide production is reduced by cell-bound SOD. At least for the chromosomal damage observed in the above-mentioned chronic inflammatory diseases with autoimmune reactions, the anticlastogenic effects of SOD may be explained by diminished superoxide production from monocytes, correlated with diminished production of clastogenic mediators. Since monocytes do not divide under the given culture conditions, it is not possible to study chromosomal damage on this cell type. Lymphocytes, which exhibited only poor SOD-labeling in comparison to monocytes, are the only dividing cells in blood cultures and those showing the chromosomal aberrations in metaphases obtained by stimulation with the mitogen phytohemagglutinin. One may ask whether the reduced aberration rate observed in lymphocytes after incubation of the cells with SOD is due only to diminished production of clastogenic compounds by monocytes in the culture system or whether the even small amount of SOD bound to the surface of lymphocytes represents an additional protection against superoxide-mediated clastogenesis. This is probably the case, since SOD is also protective in fibroblast cultures, in which other superoxide producing cells are not present. However, labeling with FITC–SOD was significantly more important for fibroblasts than for lymphocytes. Whether the protective effects are exerted by membrane-bound SOD or whether intracellular uptake is necessary for anticlastogenesis remains an open question. Whereas membrane binding of native SOD is rapid and visible after 15 min on monocytes and hepatocytes (27), its passage through the membrane appears to be relatively slow and variable according to cell type and enzyme properties. In our experiments, the cells had been pretreated with SOD during 1 h, a duration not sufficient for intracellular penetration of the enzyme according to confocal microscopy. Another question is whether internalized SOD acts at the nuclear level or in the cytoplasm. No FITC fluorescence was detected by us on the nuclei in agreement with Petkau (36), who studied uptake of radioactive SOD in different cellular fractions and found only minor quantities in the nuclear fraction.
In conclusion, our data indicate that anticlastogenic effects of SOD are not limited to the destruction of superoxide radicals generated extracellularly or leaking out of the cell, but that exogenous SOD, after binding to the cell surface, diminishes superoxide production by cells and the release of clastogenic metabolites generated via membrane-mediated mechanisms. The observation of cellular binding of SOD is not only of interest for the prevention of genotoxic effects by CF and xenotropic clastogens, acting via the intermediacy of superoxide. It is also important from a therapeutic standpoint, in particular in the above-mentioned chronic inflammatory diseases, in which oxyradical production by activated monocytes/macrophages contributes to tissue injury. Finally, since binding of the enzyme to the cells was demonstrated after 15 min, this could explain why clinical effects last for weeks and even months (37) despite the rapid clearance of injected SOD from the blood stream.
Footnotes
Abbreviations: SOD, superoxide dismutase; FITC, fluorescein isothiocyanate; PMA, phorbol 12-myristate 13-acetate; ITP, inosine triphosphate; CF, clastogenic factor(s); PMN, polymorphonuclear neutrophil.
References
- 1.Nordenson I, Beckman G, Beckman L. Hereditas. 1976;82:125–126. doi: 10.1111/j.1601-5223.1976.tb01546.x. [DOI] [PubMed] [Google Scholar]
- 2.Nordenson I. Hereditas. 1977;86:147–150. doi: 10.1111/j.1601-5223.1977.tb01223.x. [DOI] [PubMed] [Google Scholar]
- 3.Brawn K, Fridovich I. Acta Physiol Scand Suppl. 1980;492:9–18. [PubMed] [Google Scholar]
- 4.Mello-Filho A C, Meneghini R. Biochim Biophys Acta. 1984;781:56–63. doi: 10.1016/0167-4781(84)90123-4. [DOI] [PubMed] [Google Scholar]
- 5.Ochi T, Cerutti P. Proc Natl Acad Sci USA. 1987;84:990–994. doi: 10.1073/pnas.84.4.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blakely W F, Fuciarelli A F, Wegher B J, Dizdaroglu M. Radiat Res. 1990;21:338–343. [PubMed] [Google Scholar]
- 7.Emerit I, Michelson A M. Acta Physiol Scand Suppl. 1980;492:59–65. [PubMed] [Google Scholar]
- 8.Emerit I. Free Radical Biol Med. 1994;16:99–109. doi: 10.1016/0891-5849(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 9.Emerit I. In: Modern Aging Research: Free Radicals, Aging and Degenerative Diseases. Johnson J E, editor. New York: Liss; 1986. pp. 307–324. [Google Scholar]
- 10.Emerit I, Cerutti P. Proc Natl Acad Sci USA. 1981;78:1868–1872. doi: 10.1073/pnas.78.3.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emerit I, Keck M, Levy A, Feingold J, Michelson M. Mutat Res. 1982;103:165–172. doi: 10.1016/0165-7992(82)90024-0. [DOI] [PubMed] [Google Scholar]
- 12.Emerit I, Cerutti P. Nature (London) 1981;293:144–146. doi: 10.1038/293144a0. [DOI] [PubMed] [Google Scholar]
- 13.Emerit I, Levy A, Cerutti P. Mutat Res. 1983;110:327–335. doi: 10.1016/0027-5107(83)90149-5. [DOI] [PubMed] [Google Scholar]
- 14.Goh K O, Sumner H. Radiat Res. 1968;6:51–60. [PubMed] [Google Scholar]
- 15.Hollowell J G, Littlefield L G. Proc Soc Exp Biol Med. 1968;129:240–244. doi: 10.3181/00379727-129-33295. [DOI] [PubMed] [Google Scholar]
- 16.Emerit I, Levy A, Pagano G, Pinto L, Calzone R, Zatterale A. Hum Genet. 1995;96:14–20. doi: 10.1007/BF00214180. [DOI] [PubMed] [Google Scholar]
- 17.Emerit I, Khan S H, Cerutti P. Free Radical Biol Med. 1985;1:51–57. doi: 10.1016/0748-5514(85)90029-7. [DOI] [PubMed] [Google Scholar]
- 18.Khan S H, Emerit I. Free Radical Biol Med. 1985;1:443–449. doi: 10.1016/0748-5514(85)90159-x. [DOI] [PubMed] [Google Scholar]
- 19.Emerit I, Khan S H, Esterbauer H. Free Radical Biol Med. 1991;10:371–377. doi: 10.1016/0891-5849(91)90045-5. [DOI] [PubMed] [Google Scholar]
- 20.Auclair C, Gouyette A, Levy A, Emerit I. Arch Biochem Biophys. 1990;278:238–244. doi: 10.1016/0003-9861(90)90253-u. [DOI] [PubMed] [Google Scholar]
- 21.Emerit I, Levy A, Khan S H. In: Free Radicals, Lipoproteins, and Membrane Lipids. Crastes de Paulet A, Douste-Blazy L, Paoletti R, editors. New York: Plenum; 1990. pp. 99–104. [Google Scholar]
- 22.Emerit I. Methods Enzymol. 1990;186:555–564. doi: 10.1016/0076-6879(90)86150-t. [DOI] [PubMed] [Google Scholar]
- 23.Flohe L, Otting F. Methods Enzymol. 1984;105:93–104. doi: 10.1016/s0076-6879(84)05013-8. [DOI] [PubMed] [Google Scholar]
- 24.Johnston B. Methods Enzymol. 1984;105:215–220. doi: 10.1016/s0076-6879(84)05028-x. [DOI] [PubMed] [Google Scholar]
- 25.Baret A, Emerit I. Mutat Res. 1983;121:293–297. doi: 10.1016/0165-7992(83)90217-8. [DOI] [PubMed] [Google Scholar]
- 26.Osheroff N, Shelton E R, Brutlag D L. J Biol Chem. 1983;258:9536–9543. [PubMed] [Google Scholar]
- 27.Michelson A M, Puget K. Acta Physiol Scand Suppl. 1980;492:67–80. [PubMed] [Google Scholar]
- 28.Ando Y, Inoue M, Utsumi T, Morino Y, Araki S. FEBS Lett. 1988;40:216–220. doi: 10.1016/0014-5793(88)80371-5. [DOI] [PubMed] [Google Scholar]
- 29.Dini L, Rotilio G. Biochem Biophys Res Commun. 1989;162:940–944. doi: 10.1016/0006-291x(89)90763-8. [DOI] [PubMed] [Google Scholar]
- 30.Beckman J S, Minor R L, White C W, Repine J E, Rosen G M, Freeman B A. J Biol Chem. 1988;263:6884–6892. [PubMed] [Google Scholar]
- 31.Nakae D, Yoshiji H, Amanuma T, Kinusaga T, Farber J L, Konishi Y. Arch Biochem Biophys. 1990;279:315–319. doi: 10.1016/0003-9861(90)90497-m. [DOI] [PubMed] [Google Scholar]
- 32.Kyle M E, Nakae D, Sakaida I, Miccadei S, Farber J L. J Biol Chem. 1988;263:3784–3789. [PubMed] [Google Scholar]
- 33.Emerit I, Cerutti P. Carcinogenesis. 1983;4:1313–1316. doi: 10.1093/carcin/4.10.1313. [DOI] [PubMed] [Google Scholar]
- 34.Pogrebniak H W, Matthews W A, Pass H I. Surg Forum. 1990;16:101–105. [Google Scholar]
- 35.Kozumbo W J, Muehlematter D, Jörg A, Emerit I, Cerutti P. Carcinogenesis. 1987;8:521–526. doi: 10.1093/carcin/8.4.521. [DOI] [PubMed] [Google Scholar]
- 36.Petkau A, Chelack W S, Kelly K, Friesen H K. In: Pathology of Oxygen. Autor A, editor. New York: Academic; 1982. pp. 223–243. [Google Scholar]
- 37.Flohé L. Mol Cell Biochem. 1988;84:123–131. doi: 10.1007/BF00421046. [DOI] [PubMed] [Google Scholar]