

Abstract
Small ligand–receptor interactions underlie many fundamental processes in biology and form the basis for pharmacological intervention of human diseases in medicine. We report herein a genetic system, named the yeast three-hybrid system, for detecting ligand–receptor interactions in vivo. This system is adapted from the yeast two-hybrid system with which a third synthetic hybrid ligand is combined. The feasibility of this system was demonstrated using as the hybrid ligand a heterodimer of covalently linked dexamethasone and FK506. Yeast expressing fusion proteins of the hormone binding domain of the rat glucocorticoid receptor fused to the LexA DNA-binding domain and of FKBP12 fused to a transcriptional activation domain activated reporter genes when plated on medium containing the dexamethasone–FK506 heterodimer. The reporter gene activation is completely abrogated in a competitive manner by the presence of excess FK506. Using this system, we screened a Jurkat cDNA library fused to the transcriptional activation domain in yeast expressing the hormone binding domain of rat glucocorticoid receptor–LexA DNA binding domain fusion protein in the presence of dexamethasone–FK506 heterodimer. We isolated overlapping clones of human FKBP12. These results demonstrate that the three-hybrid system can be used to discover receptors for small ligands and to screen for new ligands to known receptors.
The use of small organic ligands as probes in biology has been fruitful in elucidating mechanisms of signal transduction and cell regulation (1–4). The identification of receptors for small ligands has relied primarily on in vitro biochemical methods including photocrosslinking, radiolabeled-ligand binding, and affinity chromatography (5). These methods are often laborious and time-consuming, requiring purification of sufficient quantities of target receptors from complex mixtures of cellular proteins, peptide sequencing, and subsequent cloning of the cDNAs coding for the receptors. Once a receptor has been identified and proven to be involved in the pathogenesis of disease, it becomes important to screen for small ligands that bind to and modulate the function of the receptor. These ligands then become candidates for drug development (6). Although it is possible to establish high-throughput screening assays for most receptors in vitro, it often requires the expression and purification of sufficient quantities of recombinant receptor proteins and finding a suitable readout. Many of the lead compounds identified in such screening assays are often not useful due to lack of either cell permeability or stability inside the cells. To date, there are no general methods that allow for the identification of new ligands for a specific receptor in vivo.
We report herein a general in vivo assay system that can be applied to rapidly discover receptors for known ligands and to screen for new ligands for known receptors. It is made possible by combining synthetic organic chemistry to generate heterodimeric hybrid ligands (7) with yeast genetics—i.e., the yeast two-hybrid system (8, 9). Since its development (8), the yeast two-hybrid system has proven to be a powerful tool for detecting protein–protein interactions. On a different front, it was elegantly shown that dimers of organic ligands can cause dimerization or oligomerization of two separate proteins fused to receptors to the ligands, serving as agonists to various signaling events in mammalian cells (7). To make the yeast two-hybrid system usable for studying small ligand–receptor interactions, we envisioned the use of synthetic heterodimer of two different small organic ligands as a third hybrid molecules to bring into proximity the DNA-binding domain fused to the receptor for one ligand and the activation domain fused to the receptor for a second ligand, thus activating reporter genes. This system, which we call the yeast three-hybrid system, can be applied to directly identify cDNAs encoding receptors of a ligand of interest and to screen for new ligands that bind to a specific receptor.
MATERIALS AND METHODS
Materials.
All chemical regents used in organic synthesis were purchased from Aldrich. Dexamethasone was purchased from Research Biochemicals (Natick, MA). FK506 was a gift from Fujisawa Pharmaceuticals (Osaka). The vectors and the yeast strain for the yeast two-hybrid system was provided by Roger Brent (Massachusetts General Hospital). The Jurkat cDNA library subcloned in pJG4-5 was from Brian Seed (Massachusetts General Hospital). The plasmids p6RGR and p6RGR-C656G encoding the rat glucocorticoid receptor and its mutant were provided by Keith Yamamoto (University of California, San Francisco).
Synthesis of Dexamethasone–FK506 Heterodimer.
As shown in Fig. 1, the conversion of dexamethasone into its N-hydroxysuccinimide ester (compound 1) was as described (10). The primary amine of dexamethasone (compound 2) was synthesized as follows. To a solution of dexamethasone ester (1) in tetrahydrofuran was added 4 molar equivalents of 1,10-diaminodecane and the reaction mixture was stirred at room temperature for 1 hr. The reaction mixture was concentrated in vacuo and purified by preparative thin layer chromatography (using a solvent system consisting of 89% CHCl3, 10% CH3OH, and 1% Et3N) to yield the dexamethasone primary amine (compound 2) as a white solid in 90% yield. Synthesis of the FK506 mixed carbonate (compound 3) is as described (11). Dexamethasone–FK506 heterodimer (compound 4) was synthesized as follows. Two equivalents of compound 2 and one equivalent of compound 3 were added into a solution of tetrahydrofuran containing triethylamine (excess). The mixture was stirred at room temperature for 2 hr. The reaction mixture was treated with saturated NaHCO3, extracted with EtOAc, dried by filtration through MgSO4, and concentrated in vacuo. The crude product was dissolved in acetonitrile and treated with hydrofluoric acid (49%) at room temperature for 24 hr. The reaction mixture was treated with saturated NaHCO3, extracted with EtOAc, dried by filtration through MgSO4, and concentrated in vacuo. The product was purified by preparative thin layer chromatography using EtOAc as a solvent to yield the dexamethasone–FK506 hybrid “bait” ligand in 64% combined yield for two steps. All compounds were characterized by 1H-NMR. The structure of heterodimer 4 were confirmed by 13C-NMR and high-resolution mass spectrometry in addition to 1H-NMR.
Figure 1.
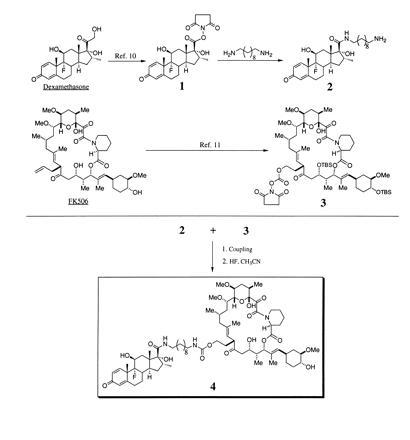
Synthesis and structure of the dexamethasone–FK506 hybrid ligand (4).
Construction of the Hook and the Prey Expression Vectors.
The hormone-binding domain-containing DNA fragments were PCR-amplified using as templates plasmids p6RGR and p6RGR-C656G, which encode full-length wild-type rat glucocorticoid receptor and the C656G mutant, respectively (12, 13). The PCR primers used to generate the vectors were 5′-GCATCAATTGGCAGGAGTCTCACAAGACCG-3′ (5′ primer starting at amino acid 524), 5′-GCATCAATTGTATCGGAAATGTCTTCAGGCT-3′ (5′ primer starting at amino acid 497), 5′-GCATCTCGAGTCATTTTTGATGAAACAG-3′ (3′ primer), and 5′-TCATGGATGTCTCTCATGGCA-3′ (F620S mutagenic primer). For constructs pEGHBD1-3, DNA fragments containing the wild-type hormone binding domain and C656G mutant were amplified using p6RGR (for pEGHBD1) and p6RGR-C656G (for pEGHBD2-3) as templates with appropriate primers to generate PCR products flanked by MfeI and XhoI sites. These fragments were digested and subcloned into the EcoRI–XhoI sites of pEG202. For constructs pEGHBD4-7, the F620S mutation was introduced using the mutagenic primer by a two-step PCR protocol (14). PCR fragments were subcloned into pEG202 similar to constructs pEGHBD1-3. The prey vector pJGFKBP was constructed by subcloning the PCR amplified human FKBP12 containing DNA fragment into pJG4-5 such that FKBP12 is in-frame with the activation domain. The DNA inserts in all plasmids were confirmed by sequencing.
Three-Hybrid Screen for FK506-Binding Proteins.
The yeast strain EGY48ura3 trp1 his3 lexA operator-LEU2 was transformed with pEGHBD7 and pSH18-34 and plated onto synthetic complete (SC) medium (His−, Ura−). The resultant EGY48 harboring pEGHBD7 and pSH18-34 was transformed with a Jurkat cDNA library subcloned into pJG4-5 (15). The transformed yeast cells (1.62 × 106 cells) were plated onto SC medium (pH 6.5, His−, Ura−, Trp−, Leu−) containing galactose and 1 μM dexamethasone–FK506. A total of 101 colonies were collected between days 4 and 10. All colonies were plated onto SC medium (His−, Ura−, Trp−, Leu−) containing galactose. A total of 92 colonies displayed “bait”-independent growth. The remaining 9 colonies were plated onto SC medium (pH 6.5, His−, Ura−, Trp−, Leu−) containing galactose and 1 μM dexamethasone–FK506 in the presence of 10 μM FK506. The growth of three colonies could be completely inhibited by FK506. The “fish” plasmids were retrieved from these three yeast strains (16) and transformed into Escherichia coli DH5α for preparation of the plasmids. The DNA inserts in these plasmids were sequenced by an Applied Biosystems automated sequencer at the Massachusetts Institute of Technology Biopolymers Laboratory.
RESULTS AND DISCUSSION
The concept and the components of the yeast three-hybrid system are shown schematically in Fig. 2. This system utilizes three hybrid molecules to reconstitute transcription of reporter genes in a manner similar to the two-hybrid system (8, 9). The three hybrid molecules consist of a hybrid fusion protein designated the “hook,” a second hybrid fusion protein designated the “fish,” and a synthetic hybrid ligand designated the “bait.” The hybrid ligand is a covalently linked heterodimer of two small ligands, A and B. If ligand A binds to its receptor fused to a DNA-binding domain (i.e., the “hook”) and ligand B binds to its receptor fused to a transcriptional activation domain (i.e., the “fish”), reporter genes will be activated, allowing for the selection of yeast cells that harbor the relevant receptors. It has been demonstrated that dimers of small organic ligands, termed chemical inducers of dimerization, can be used to induce dimerization or oligomerization of cellular proteins tagged with receptors for the dimeric ligands in mammalian cells, although fusions of multiple copies of receptors to the targeted protein are required for optimal response (7, 17). We reasoned that the three-hybrid system, which combines a chemically covalent linked heterodimer of two organic ligands with the yeast two-hybrid system, would work provided that the affinity of the hybrid ligand to the receptors are sufficiently high to allow detectable reporter gene activation when a single copy of the “hook” and the “fish” receptors is present in the hybrid fusion proteins.
Figure 2.
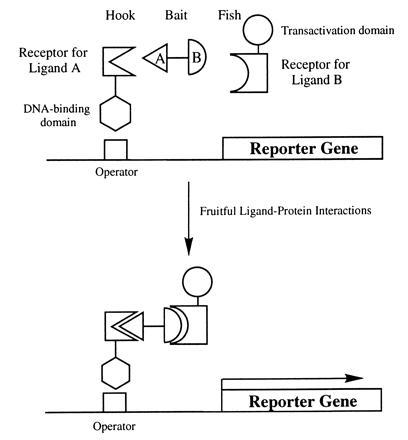
Diagrammatic representation of the components of the yeast three-hybrid system. The synthetic “bait” hybrid ligand consists of ligand A (triangle) and ligand B (semicircle) connected by a linker. The “hook” fusion protein consists of the receptor for ligand A fused to the DNA-binding domain of a transcription factor. The “fish” fusion protein consists of the receptor for ligand B fused to a transactivation domain of a transcription factor. The relative sizes of the three hybrid molecules are not drawn to scale. The “hook,” the “bait,” and the “fish” form a trimeric complex, and the resulting proximity of the transactivation domain and the DNA-binding domain leads to activation of the reporter gene.
To test the feasibility of the yeast three-hybrid system, we synthesized a hybrid ligand from the steroid hormone agonist dexamethasone and the immunosuppressant FK506. We chose this hybrid ligand since an extensive amount of structure–activity data was available on the binding of both ligands to their respective receptors. Dexamethasone is known to bind the glucocorticoid receptor thereby modulating gene expression (18) and FK506 is known to bind FK506 binding proteins (FKBPs) (3). Both dexamethasone and FK506 have been modified into affinity reagents for the isolation and identification of their receptors (10, 19). Homodimers of FK506 have also been made and shown to induce oligomerization of proteins fused to FKBPs (7). Taking advantage of the structure–activity information, we synthesized the dexamethasone–FK506 heterodimer by covalently linking the two molecules with a 10-carbon tether through positions on each ligand that when modified do not abrogate receptor binding (Fig. 1).
We used the yeast two-hybrid system based on the LexA DNA-binding domain and the transactivation domain from the bacterial protein B42 (9). We constructed vectors encoding the “hook” hybrid protein composed of the LexA DNA-binding domain fused to the hormone-binding domain of the rat glucocorticoid receptor (rGR). To optimize the interaction between the dexamethasone portion of the hybrid ligand and the hormone-binding domain of rGR, several variants of the “hook” fusion protein were generated (Fig. 3A, pEGHBD1-7). In addition to varying the length of the hormone binding domain fused to LexA, we incorporated two point mutations. C656G, the first of these mutations, is known to increase the affinity of dexamethasone for the hormone binding domain (12). F620S, the second mutation, is known to increase the availability of rGR that is capable of interacting with dexamethasone in yeast (13). We also constructed two double mutants, pEGHBD6 and pEGHBD7, that are expected to have even higher affinity for dexamethasone. For the “fish” hybrid protein, we constructed a vector (pJG-FKBP12) encoding the human FKBP12 fused to the transcriptional activator B42 (9).
Figure 3.
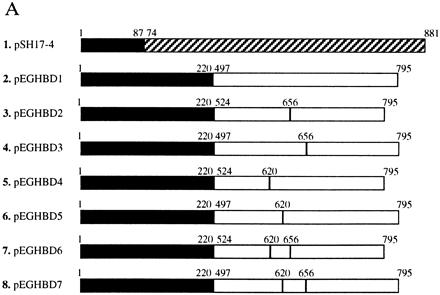
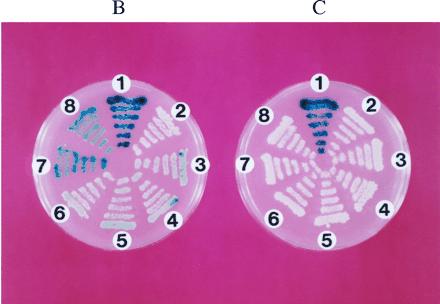
(A) Control fusion protein (pSH17-4) between LexA DNA-binding domain (solid bars) and Gal4 transactivation domain (shaded bars) and variants of “hook” fusion proteins between LexA DNA-binding domain and hormone-binding domain of rGR (open bars). The first and the last amino acid of the LexA DNA binding protein, Gal4 transactivation domain, and the hormone binding domain of rGR, as well as the relative position of the two mutations in the hormone-binding domain of rGR are indicated above each construct. (B) Activation of the lacZ reporter gene by dexamethasone–FK506 hybrid ligand. EGY48 transformed with different “hook” fusion protein constructs (numbered as in A), the pJG-FKBP12 “fish” fusion protein construct and the reporter pSH18-34 from a master plate were replica plated onto SC medium (His−, Ura−, Trp−) containing galactose, 5-bromo-4-chloro-3-indolyl β-d-galactoside, and 1 μM dexamethasone–FK506 and incubated at 30°C for 3 days. (C) Inhibition of the lacZ reporter gene activation by FK506 competition. All conditions and strains of yeast are identical to those in B except that 10 μM of FK506 was present in the medium.
The yeast strain EGY48 was transformed with different “hook” vectors along with the FKBP-encoding “fish” vector and the reporter vector pSH18-34 (20). The transformed yeast strains were plated onto SC medium (His−, Ura−, Trp−) containing galactose and 1 μM of the dexamethasone–FK506 hybrid ligand. Indeed, activation of the lacZ gene was observed for most of the yeast transformants (Fig. 3B). As expected, the level of activation of the lacZ gene correlated with the overall affinity of the different hormone binding domain variants. While no detectable activation of lacZ gene was observed for yeast expressing the wild-type hormone binding domain of rGR, the strongest activation was observed in yeast expressing the “hook” fusion proteins that contain both C656G and F620S mutations (Fig. 3 A and B). When the same yeast were replica-plated on SC medium (His−, Ura−, Trp−) containing galactose, 1 μM dexamethasone–FK506 hybrid ligand, and 10 μM FK506, activation of the lacZ gene was completely abolished (Fig. 3C). This indicated that FK506 was able to compete for the FKBP12 “fish” protein, preventing the dexamethasone–FK506 hybrid ligand from activating the reporter gene.
To determine whether the yeast three-hybrid system could be used to identify receptors for a ligand, a screen for FK506 binding proteins was carried out using the “hook” hybrid protein that displayed the strongest activation of the lacZ reporter gene (Fig. 3A, pEGHBD6). We transformed EGY48 with pEGHBD6, pSH18-34, and pJG4-5 containing a Jurkat cDNA library. The transformants were plated onto SC medium (His−, Ura−, Trp−, Leu−) containing galactose and the dexamethasone–FK506 hybrid ligand. Of the colonies that grew on leucine deficient medium, most displayed growth independent of dexamethasone–FK506 heterodimer. When those that displayed growth dependence on the dexamethasone–FK506 hybrid ligand were plated onto SC medium (His−, Ura−, Trp−, Leu−) containing galactose, dexamethasone–FK506 (1 μM), and an excess amount of FK506 (10 μM), growth of three colonies was completely inhibited by FK506. We retrieved the “fish” vectors from the FK506-sensitive colonies and sequenced the cDNA inserts. These inserts contained two variants of human FKBP12 (one of which appeared twice) with different 5′ start sites (Fig. 4). It took less than 3 weeks to isolate and identify the FKBP12 cDNAs using the yeast three-hybrid system. In comparison, it would have taken months if not a year to accomplish the same task using biochemical methods that require the isolation of sufficient quantities of FKBP, determination of peptide sequences and cloning of the cDNA. This demonstrates that the yeast three-hybrid system is a powerful method to identify receptors for a ligand.
Figure 4.
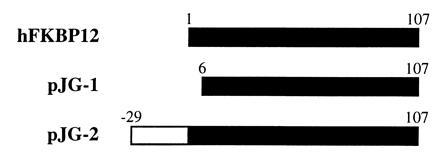
FKBP12 cDNA variants isolated through the yeast three-hybrid screen of a Jurkat cDNA library. pJG-1 encodes FKBP12 minus the first 5 amino acids; pJG-2, isolated in duplicate, encodes FKBP12 with an extended 29-amino acid fragment at its amino terminus.
Multiple isoforms of FKBPs exist in Jurkat T cells (21). FKBP12 isolated through the yeast three-hybrid screen is the most abundant isoform and has the highest affinity for FK506, accounting for its identification as the first member of the FKBP immunophilin family by biochemical methods (19, 22). That other FKBPs are not identified with the three-hybrid screen may be attributed to the fact that yeast expresses high levels of FKBPs that compete for the dexamethasone–FK506 hybrid ligand (23, 24), decreasing the sensitivity of this particular screen. It will be interesting to see whether other receptor families with lower abundance in yeast can be identified more efficiently.
A number of factors affecting yeast three-hybrid interactions remain to be determined. First of all, it has been estimated that the yeast two-hybrid system can detect protein interactions with affinities greater than 1 μM (25). But it is not known what type of affinity is required for a detectable three-hybrid interaction between a ligand and its receptor. The fact that wild-type hormone binding domain of rGR with a Kd of 5 nM (12) did not give detectable activation of lacZ gene (Fig. 3) seems to suggest affinities in the nanomolar or subnanomolar range for at least one ligand–receptor pair of the three-hybrid system. But this is complicated, in part, by the aforementioned competition by yeast FKBPs for the dexamethasone–FK506 hybrid ligand that decreases its availability for three-hybrid interactions. A systematic study using ligand–receptor pairs with different affinities will be needed to fully assess the minimum affinity requirement for the three-hybrid system. Second, as dexamethasone and FK506 are both hydrophobic drugs, it is not surprising that the corresponding hybrid ligand was able to diffuse into yeast cells. To make the three-hybrid system useful for a wide range of ligands including those that are hydrophilic or charged, it will be important to generate yeast strains that are more permeable without significantly affecting yeast viability. Third, like the yeast two-hybrid system, the three-hybrid system is not easily applicable to most membrane-associated proteins, limiting the types of receptors to soluble proteins or soluble domains of membrane-associated proteins. It is imaginable that membrane protein-based two- and three-hybrid systems would further expand the utility of the current systems.
Despite some of the limitations, the yeast three-hybrid system offers considerable advantages over classical biochemical methods (5) for identifying receptors for small ligands with the main advantage being the direct isolation and identification of cDNAs coding for receptors. The ease with which a large number of yeast colonies can be manipulated makes the three-hybrid system ideal for structure–function analysis of receptors to identify residues that are important for ligand recognition in conjunction with methods such as random mutagenesis. It will also be useful for the generation of high-affinity binding proteins to small ligands such as environmentally hazardous compounds or drugs; these proteins may be useful as detection and diagnostic reagents.
The three-hybrid system can also be applied to screen for novel ligands for a given receptor both in yeast and in mammalian cells (26, 27) in vivo. The FK506 competition experiment (Fig. 3C) demonstrates that this system can be used to identify ligands that compete for a known ligand–receptor interaction. With the availability of synthetic combinatorial libraries of small organic molecules (28), this system offers a highly efficient way of identifying such ligands. In the case of a receptor with no known ligand, it is conceivable that a hybrid combinatorial library covalently linked to a known ligand such as dexamethasone can be screened to discover new lead compounds for drug development. Unlike the constrained or linear peptide libraries in the yeast two-hybrid system (29, 30) or the bacteriophage display system (31–33), the types of ligands compatible with the yeast three-hybrid system are not confined to genetically encoded peptides; they can be any type of ligand including peptides composed of d-amino acids, peptidomimetics, or any synthetic organic compounds. Compared with screening assays in vitro, the ligands that are identified through the three-hybrid system are already selected for their permeability into eukaryotic cells and their stability in a cellular environment, thereby increasing the efficiency of the screening. There is thus great potential for the use of three-hybrid technology in drug discovery.
Acknowledgments
We thank Drs. Roger Brent, Keith Yamamoto, and Brian Seed for plasmids, yeast strains, and the Jurkat cDNA library; Fujisawa Pharmaceutical (Osaka) for FK506. We thank Dr. Chris Kaiser for helpful discussions. We thank Chris Armstrong, Luo Sun, and Eddy Chen for technical help. We are grateful to Drs. Harvey Lodish, Carl Pabo, Hidde Ploegh, Richard Hynes, Christine Loh, Paul Weinreb, and Mr. Ben Turk for comments on the manuscript. This work was supported in part by the Massachusetts Institute of Technology, the National Cancer Institute (through a cancer training grant to E.J.L.), the Chicago Community Trust, and the Rita Allen Foundation. J.O.L. is a Pfizer–Laubach Career Development Assistant Professor.
Footnotes
Abbreviations: rGR, rat glucocorticoid receptor; FKBP, FK506 binding protein.
References
- 1.Yamamoto K R. Annu Rev Genet. 1985;19:209–252. doi: 10.1146/annurev.ge.19.120185.001233. [DOI] [PubMed] [Google Scholar]
- 2.Evans R M. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schreiber S L. Science. 1991;251:283–287. doi: 10.1126/science.1702904. [DOI] [PubMed] [Google Scholar]
- 4.Klausner R D, Donaldson J G, Lippincott-Schwartz J. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jakoby W B, Wilchek M. Methods Enzymol. 1974;46:1–196. doi: 10.1016/s0076-6879(74)34004-9. [DOI] [PubMed] [Google Scholar]
- 6.Hardman J G, Limbird L E, Molinoff P B, Ruddon R W, Gilman A G. The Pharmacological Basis of Therapeutics. New York: McGraw–Hill; 1996. [Google Scholar]
- 7.Spencer D M, Wandless T J, Schreiber S L, Crabtree G R. Science. 1993;262:1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- 8.Fields S, Song O. Nature (London) 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 9.Gyuris J, Golemis E, Chertkov H, Brent R. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- 10.Manz B, Heubner A, Kohler I, Grill H-J, Pollow K. Eur J Biochem. 1983;131:333–338. doi: 10.1111/j.1432-1033.1983.tb07266.x. [DOI] [PubMed] [Google Scholar]
- 11.Pruschy M N, Spencer D M, Kapoor T M, Miyake H, Crabtree G R, Schreiber S L. Chem Biol. 1994;1:163–172. doi: 10.1016/1074-5521(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 12.Chakraborti P K, Garabedian M J, Yamamoto K R, Simons S S., Jr J Biol Chem. 1991;266:22075–22078. [PubMed] [Google Scholar]
- 13.Garabedian M J, Yamamoto K R. Mol Biol Cell. 1992;3:1245–1257. doi: 10.1091/mbc.3.11.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarker G, Sommer S S. BioTechniques. 1990;8:404–407. [PubMed] [Google Scholar]
- 15.Gietz D, St. Jean A, Woods R A, Schiestl R H. Nucleic Acids Res. 1992;20:1245. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current Protocol in Molecular Biology. New York: Greene & Wiley; 1995. [Google Scholar]
- 17.Austin D J, Crabtree G R, Schreiber S L. Chem Biol. 1994;1:131–136. doi: 10.1016/1074-5521(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 18.Cohen J J. In: Anti-Inflammatory Steroid Action, Basic, and Clinical Aspects. Schleimer R P, Claman H N, Oronsky A L, editors. San Diego: Academic; 1989. pp. 111–131. [Google Scholar]
- 19.Harding M W, Galat A, Uehling D E, Schreiber S L. Nature (London) 1989;341:758–760. doi: 10.1038/341758a0. [DOI] [PubMed] [Google Scholar]
- 20.Golemis E, Brent R. Mol Cell Biol. 1992;12:3006–3014. doi: 10.1128/mcb.12.7.3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fretz H, Albers M W, Galat A, Standaert R F, Lane W S, Burakoff S J, Bierer B E, Schreiber S L. J Am Chem Soc. 1993;113:1409–1411. [Google Scholar]
- 22.Siekierka J J, Hung S H Y, Poe M, Lin C S, Sigal N H. Nature (London) 1989;341:755–757. doi: 10.1038/341755a0. [DOI] [PubMed] [Google Scholar]
- 23.Heitman J, Movva N R, Hiestand P C, Hall M N. Proc Natl Acad Sci USA. 1991;88:1948–1952. doi: 10.1073/pnas.88.5.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiederrecht G, Brizuela L, Elliston K, Sigal N H, Siekierka J J. Proc Natl Acad Sci USA. 1991;88:1029–1033. doi: 10.1073/pnas.88.3.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Estojak J, Brent R, Golemis E. Mol Cell Biol. 1995;15:5820–5829. doi: 10.1128/mcb.15.10.5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang C V, Barrett J, Villa-Garcia M, Resar L M S, Kato G J, Fearon E R. Mol Cell Biol. 1991;11:954–962. doi: 10.1128/mcb.11.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belshaw P J, Ho S N, Crabtree G R, Schreiber S L. Proc Natl Acad Sci USA. 1996;93:4604–4607. doi: 10.1073/pnas.93.10.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gallop M A, Barrett R W, Dower W J, Fodor S P A, Gordon E M. J Med Chem. 1994;37:1233–1251. doi: 10.1021/jm00035a001. [DOI] [PubMed] [Google Scholar]
- 29.Yang M, Wu Z, Fields S. Nucleic Acids Res. 1995;23:1152–1156. doi: 10.1093/nar/23.7.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colas P, Cohen B, Jessen T, Grishina I, McCoy J, Brent R. Nature (London) 1996;380:548–550. doi: 10.1038/380548a0. [DOI] [PubMed] [Google Scholar]
- 31.Scott J K, Smith G. Science. 1990;249:386–390. doi: 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- 32.Devlin J J, Panganiban L C, Devlin P E. Science. 1990;249:404–406. doi: 10.1126/science.2143033. [DOI] [PubMed] [Google Scholar]
- 33.Cwirla S E, Peters E A, Barrett R W, Dower W J. Proc Natl Acad Sci USA. 1990;87:6378–6382. doi: 10.1073/pnas.87.16.6378. [DOI] [PMC free article] [PubMed] [Google Scholar]