

Abstract
To further elucidate the mechanism and dynamics of bacteriophage T4 holoenzyme formation, a mutant polymerase in which the last six carboxyl-terminal amino acids are deleted, was constructed, overexpressed, and purified to homogeneity. The mutant polymerase, designated ΔC6 exo−, is identical to wild-type exo− polymerase with respect to kcat, kpol, and dissociation constants for nucleotide and DNA substrate. However, unlike wild-type exo− polymerase, the ΔC6 exo− polymerase is unable to interact with the 45 protein to form the stable holoenzyme. A synthetic polypeptide corresponding to the carboxyl terminus of the wild-type exo− polymerase was tested as an in vitro inhibitor of bacteriophage T4 DNA replication. Surprisingly, the peptide does not directly inhibit holoenzyme complex formation by disrupting the interaction of the polymerase with the 45 protein. On the contrary, the peptide appears to disrupt the interaction of the 44/62 protein with the 45 protein, suggesting that the 44/62 protein and the polymerase use the same site on the 45 protein for functional interactions. Data presented are discussed in terms of a model correlating the functionality of the carboxyl terminus of the polymerase for productive interactions with the 45 protein as well as in terms of the 45 protein concomitantly interacting with the 44/62 protein and polymerase.
Keywords: DNA replication, accessory proteins, holoenzyme, peptides, rational drug design
The bacteriophage T4 holoenzyme is a complex of the T4 DNA polymerase and 45 protein, assembled onto duplex DNA in an ATP-dependent process by the 44/62 protein (reviewed in refs. 1 and 2). Although intimately involved in the assembly process, the 44/62 protein does not remain part of the holoenzyme complex but instead acts catalytically (3, 4). The role of the 45 protein is to act as a sliding clamp that tethers the polymerase to the DNA primer/template, increasing the processivity of the polymerase. The processivity factor of the T4 system is believed to encircle DNA in a manner identical to that of the β subunit (5) and proliferating cell nuclear antigen (6), based upon preliminary x-ray crystallographic studies of the 45 protein (J. Kuriyan, personal communication) as well as functional similarities among the three clamps. While the dynamics of clamp opening and/or closing are still speculative, it is believed that the 44/62 protein clamp loader opens and closes the 45 protein clamp by an energy transduction mechanism that closely couples the energy of ATP hydrolysis to some physical force (A.J.B. and S.J.B., unpublished data). Once loaded onto DNA, the 45 protein and polymerase form a tightly associated complex that is highly stable on duplex DNA (8). Of paramount interest and importance is the mechanism and molecular contacts describing how the sliding clamp and polymerase interact to form the stable holoenzyme.
Studies of processive DNA replication from various systems have revealed the qualitative importance of the carboxyl-terminus of the polymerase in the functional interaction with its respective accessory protein. Herpes simplex virus holoenzyme is composed of the DNA polymerase and the UL42 accessory protein. Similar to the T4 system, the accessory protein is not necessary for polymerase activity but increases the processivity of the polymerase (9). Residues within the last 160 aa of the polymerase are reported to be critical for the polymerase interacting with UL42 and are thus necessary for viral replication (10, 11). Similarly, a pseudorabies polymerase mutant lacking the 30 carboxyl-terminal amino acids also possesses polymerase activity but is deficient in interacting with its processivity factor (12). Finally, a naturally occurring mutant of the bacteriophage T4 polymerase in which 20% of the carboxyl-terminus is deleted (B22 Amber mutant) lacks polymerase activity but still possesses DNA binding and exonuclease activity (13). However, the deletion mutant does not exhibit processive exonuclease activity in the presence of the T4 accessory proteins, suggesting that the carboxyl-terminal domain of the T4 DNA polymerase may also contain residues responsible for proper interactions with the processivity factor.
Activation of bacteriophage T4 late transcription in vitro is also mediated by the T4 DNA polymerase accessory proteins, the mechanism of which is believed to be similar to that of DNA replication activation (14). In both processes, the 44/62 protein loads the 45 protein onto duplex DNA in an ATP-dependent manner. In transcriptional activation, however, the interaction of the 45 protein with the RNA polymerase is mediated by the T4-encoded 33 protein (14), which apparently contacts both the 45 protein and RNA polymerase (15). Further studies by the Geidushek group indicated that the carboxyl terminus of the 33 protein is necessary for proper interactions with the 45 protein but not with the RNA polymerase (16). Not surprisingly, the carboxyl terminus of the 33 protein and the T4 DNA polymerase are very similar, especially the last 6 aa (Fig. 1). Most likely, the carboxyl-terminal portion of both proteins play a pivotal role in the physical interaction with the 45 protein, presumably by similar mechanisms.
Figure 1.
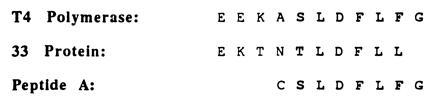
Carboxyl terminal alignment of the bacteriophage T4 DNA polymerase (gp43) and the bacteriophage T4 RNA polymerase binding protein (gp33). Amino acid sequence of the polypeptide used for inhibition studies is also shown.
Based upon these observations, the identity of molecular contacts between the 45 protein and DNA polymerase was addressed by kinetic measurements using a carboxyl terminal deletion mutant of the T4 exo− polymerase. Although the mutant polymerase displays mechanistic and kinetic properties identical to that of wild-type T4 exo− polymerase, the mutant enzyme does not interact with the 45 protein to form the holoenzyme. A peptide corresponding to the carboxyl-terminus of the polymerase inhibits the interaction of the 45 protein with the 44/62 clamp loading protein, suggesting that the 45 protein uses a common face to interact with both the 44/62 protein and polymerase.
MATERIALS AND METHODS
Materials.
[γ-32P]ATP, [α-32P]dATP, and [α-32P]dCTP were purchased from New England Nuclear. Unlabeled dNTPs (ultrapure) were obtained from Pharmacia. ATP, phosphoenol pyruvate, MgCl2, Mg(OAc)2, and all buffers were from Sigma. NADH was from Boehringer Mannheim. Synthetic polypeptides were the generous gift of Michael Moore of SmithKline Beecham. All restriction and DNA-modifying enzymes used during molecular cloning procedures were from New England Biolabs, Promega, United States Biochemical, or Boehringer Mannheim. All other materials were obtained from commercial sources and were of the highest available quality. Lactate dehydrogenase and pyruvate kinase were from Sigma. These enzymes were dialyzed against 25 mM Tris (pH 7.5) and stored in 20% glycerol at −20°C.
DNA Substrates.
All oligonucleotides, including those containing biotin derivatives, were synthesized by Operon Technologies (Alameda, CA) and purified as previously described by Capson et al. (17). Duplex DNA containing biotin derivatives (Bio-34/62/36-mer) was prepared as previously described (3, 4), while 13/20-mer and 20/27-mer duplex DNA substrates were prepared by the protocol described by Kuchta et al. (18). All duplex DNA was then purified as described by Capson et al. (17) and quantitated as described by Kuchta et al. (18).
Enzymes.
The T4 exonuclease-deficient polymerase D129A (Asp-219 → Ala mutation) was purified as previously described (19). The T4 accessory proteins 44/62 and 45 were purified from overproducing strains obtained from William Konigsberg (Yale University). The concentration of the 44/62 and 45 proteins are reported as units of 4:1 complex and trimer, respectively, in agreement with the stoichiometry reported by Jarvis et al. (20).
Mutant Polymerase Construction and Purification.
The Escherichia coli strain MV1190 (supE) used for the expression of the T4 polymerase mutant was a generous gift from Nancy Nossal (National Institutes of Health). The plasmid containing the T4 wild-type exo− polymerase gene, pTC1203, was previously constructed by Frey et al. (19). Removal of the six carboxyl terminal residues of the T4 exo− polymerase took advantage of a HindIII site located nine codons before the termination signal in pTC1203. The plasmid was constructed by ligation of three fragments isolated as follows. A 265-bp fragment was isolated by agarose gel electrophoresis after restriction digest of pTC1203 with HindIII and BstXI. Similarly, a 5280-bp fragment was obtained after restriction with PstI and BstXI. The third fragment is a small, synthetic 12/20-mer that introduces a termination code after Ser-892. The synthesized oligonucleotides are 5′-AGCTAGCTGATAGGGGTGCA and 5′-CCCCTATCAGCT. After purification and annealing, the 12/20-mer was 5′-labeled by T4 polynucleotide kinase. The three purified fragments were then ligated to yield the expected plasmid vector. After transformation in MV1190, the clone expressing the deletion mutant was identified by the introduction of a new NheI site concomitant with the removal of the unique PstI site in the new plasmid. The T4 ΔC6 exo− polymerase was purified as previously described for wild-type exo− polymerase (20).
Methods.
All assays measuring ATP hydrolysis by the 44/62 protein were as previously described (4). Assays for the measurement of holoenzyme complex formation were as described by Kaboord and Benkovic (3) and by Berdis and Benkovic (4). The amount of complex formation is equivalent to the amount of fully elongated product (62-mer; ref. 3).
All rapid quench experiments were performed at 20°C on the instrument described by Johnson (21). The assay buffer used in all kinetic studies consisted of 25 mM Tris-OAc (pH 7.5), 150 mM KOAc, and 10 mM 2-mercaptoethanol. Reactions were quenched into EDTA (0.5 M; pH 7.4) and diluted 1:1 with sequencing gel load buffer (22). Polymerization reactions were monitored by analysis of the products on 16 or 20% sequencing gels as described by Mizrahi et al. (23). Gel images were obtained with a Molecular Dynamics PhosphorImager. Product formation was quantitated by measuring the ratio of radiolabeled extended and nonextended primer. The ratios of product formation are corrected for substrate in the absence of polymerase (zero point). Corrected ratios are then multiplied by the concentration of primer/template used in each assay to yield total product. All concentration are listed as final solution concentrations.
Determination of the equilibrium dissociation constant, KD, for the 13/20-mer.
T4 ΔC exo− polymerase (75 nM) was incubated with 13/20-mer (12.5–200 nM) in assay buffer containing EDTA (100 μM) and mixed with dATP (50 μM) containing Mg-acetate (10 mM) in assay buffer. The reactions were quenched with EDTA (100 mM) at variable times (5–500 ms). Polymerization products were analyzed as described above.
Determination of the equilibrium dissociation constant, KD, for dATP.
ΔC6 exo− polymerase (75 nM) was incubated with 13/20-mer (500 nM) in assay buffer containing EDTA (100 μM) and mixed with dATP (1–50 μM) containing Mg-acetate (10 mM) in assay buffer. The reactions were quenched with EDTA (100 mM) at variable times (5–1000 ms), and the products were analyzed as described above.
Method for measuring T4–DNA dissociation rate constants.
The 13/20-mer DNA substrate (200 nM) and either 100 nM T4 exo− polymerase or 100 nM ΔC6 exo− polymerase were preincubated in one syringe and then mixed with an equal volume of single-stranded DNA (2 mg/ml). The mixture was allowed to incubate for times ranging from 5 ms to 20 s, and any polymerase–DNA complex was chased to product by the addition of Mg-acetate (10 mM) and dATP (20 μM) with a constant reaction time of 50 ms. Reactions were quenched by the addition of 100 mM EDTA after the 50-ms reaction time. The production of 14/20-mer followed a single exponential decay from which the observed rate constant was determined.
RESULTS AND DISCUSSION
The bacteriophage T4 DNA core holoenzyme is composed solely of the DNA polymerase and the 45 protein (3, 4). To establish firmly the requirements for productive interactions of the polymerase with the sliding clamp, a mutant T4 DNA polymerase lacking the last 6 aa from the carboxyl terminus was constructed, overexpressed, and purified to homogeneity. To address the role these 6 aa play in interacting with the 45 protein while forming the holoenzyme, it is necessary first to establish that the mutant polymerase possesses the same intrinsic kinetic and mechanistic properties of the wild-type exo− polymerase. These properties include binding affinity for DNA primer/template and correct nucleotide as well as the rate of polymerization utilizing defined primer/templates. The ability of the mutant polymerase to functionally interact with the 45 protein can then be unambiguously demonstrated by the established protocols for measuring holoenzyme complex formation, and definitive structure–function relationships can be derived for the process and dynamics of holoenzyme formation.
Kinetic Characterization of the ΔC6 exo− T4 Polymerase.
Pre-steady-state and steady-state kinetics of first nucleotide incorporation by the ΔC6 exo− polymerase were examined and compared with wild-type exo− polymerase. In these experiments, the enzyme is provided with only the next nucleotide for incorporation (dATP) such that the 13/20-mer substrate is converted to 14/20-mer product. The reaction was performed by mixing a solution containing either 75 or 100 nM T4 ΔC6 exo− polymerase and 500 nM 5′-labeled 13/20-mer in reaction buffer with an equal volume of a solution containing 10 mM Mg-acetate and 50 μM dATP in the same reaction buffer. The reaction was then terminated at various times by the addition of 500 mM EDTA. The preequilibration of the polymerase and DNA results in a biphasic reaction profile characterized by a rapid initial phase followed by a second, slower phase (data not shown). The burst corresponds to the conversion of all productive enzyme–DNA complexes formed during preequilibration to product, while the steady-state phase represents a slow step following chemistry that involves product release and subsequent turnover of remaining substrate. The burst amplitudes obtained in both cases were equal to the amount of enzyme added (75 and 100 nM, respectively). Both the pre- and steady-state rate constants for the ΔC6 exo- polymerase (300 s−1 and 2–3 s−1, respectively) are comparable to those obtained for the wild-type exo− polymerase [400 s−1 and 3–4 s−1, respectively (20)].
The binding affinity of the mutated polymerase for the DNA primer/template was directly assessed by two distinct independent methods. The first measurement was the determination of the equilibrium dissociation constant (KD) of the 13/20-mer from enzyme. In these experiments, the polymerization reaction was monitored using a constant amount of ΔC6 exo− polymerase (75 nM) and variable amounts of 13/20-mer (12.5–200 nM). The preincubated enzyme–DNA solution was then mixed versus a solution containing Mg-acetate (10 mM) and dATP (50 μM) and quenched at variable times with 100 mM EDTA. The observed pre-steady-state burst of single nucleotide incorporation is consistent with rapid elongation of the ΔC6 exo−:13/20-mer complex, and the amplitude of the burst is a measure of the level of this complex. For each DNA concentration, the burst amplitude was determined by fitting the corresponding data to an equation describing an initial burst followed by a steady state (data not shown). The burst amplitudes obtained from each time course were fit to the quadratic equation [ΔC6 exo−·13/20-mer] = 0.5(KD + [ΔC6 exo−] + [13/20-mer] − 0.25(KD) + [ΔC6 exo−] + 13/20-mer)2 − [ΔC6 exo−·13/20-mer]1/2 to yield KD = 10 ± 1 nM. This value is comparable to the values of 70 ± 7 nM and 40 ± 4 nM obtained for the wild-type exo+ (17) and exo− polymerase (20), respectively. The lower KD value indicates that the ΔC6 exo− polymerase binds tighter to DNA primer/template, suggesting that these 6 aa at the carboxyl terminus might normally hinder binding of the polymerase by itself to the primer/template.
The second method was to measure the rate of dissociation of 13/20-mer from the polymerase. Duplex DNA was preincubated with enzyme and mixed with an enzyme trap (2 mg of single-stranded DNA per ml) for varying incubation times followed by the immediate addition of Mg-acetate and dATP. The reaction solution was quenched with EDTA to a final concentration of 100 mM after a 50-ms reaction time. The amount of 14/20-mer formed was determined by sequencing gel electrophoresis providing data described by a decreasing single exponential at a rate of 5 ± 1 s−1 (data not shown). This value represents the rate constant for dissociation of the 13/20-mer from the ΔC6 exo− polymerase and agrees very well with the value of 3 s−1 obtained for wild-type exo+ polymerase (17). Collectively, the data indicate that the ΔC6 exo− polymerase binds to duplex DNA as tightly as either wild-type exo− or exo+ polymerase. Thus, deletion of the 6 aa from the carboxyl-terminus of the polymerase does not affect its intrinsic binding capabilities.
To verify that the mutant polymerase did not have an altered binding affinity for nucleotide substrate, the equilibrium dissociation constant (KD) of the first nucleotide to be incorporated was measured. Constant amounts of enzyme (75 nM) and DNA (500 nM) were mixed with variable quantities of dATP (1–50 μM) in the presence of 10 mM Mg-acetate and quenched at various times by the addition of 100 mM EDTA. Each time course was biphasic, and the first order rate constants from each time course were determined by computer fit to the equation describing an initial burst followed by a steady state (data not shown). These rate constants were then fit to the hyperbolic equation kobsd = kpol[dATP]/([dATP] + KD), yielding values of 8 ± 3 μM for KD(dATP) and 300 ± 40 s−1 for kpol. These values compare well to the values of 20 μM and 400 s−1 obtained for wild-type exo+ T4 polymerase (17) and 10 μM and 400 s−1 for wild-type exo− T4 polymerase (20). These data further confirm that the deletion mutant is identical kinetically to wild-type exo− polymerase. Thus, it is reasonable to presume that any negative effect on holoenzyme complex formation can clearly be attributed solely to the absence of the 6 aa.
The Carboxyl Terminus of the T4 Polymerase Is Required for Holoenzyme Complex Formation.
The formation of holoenzyme complex using the ΔC6 exo− polymerase was examined using a defined primer/template system and directly compared with the ability of the wild-type exo− polymerase to form the holoenzyme complex. The initial assay measured the decrease in the rate of ATP hydrolysis by the 44/62 protein upon formation of the holoenzyme complex as previously described (4). Normally, the ATPase activity of the 44/62 protein is synergistically activated in the presence of stoichiometric quantities of 45 protein and Bio-34/62/36-mer DNA substrate to yield a steady-state rate of ATP hydrolysis of ≈200 nM/s (Fig. 2A). However, upon the addition of a stoichiometric quantity of T4 exo− polymerase, the ATPase activity of the 44/62 protein decreases to a rate of ≈20 nM/s. The decrease in ATP hydrolysis by the 44/62 protein under these conditions is indicative of holoenzyme complex formation (4). The identical assay using a stoichiometric quantity of ΔC6 exo− polymerase instead of wild-type exo− polymerase showed no decrease in the ATPase activity of the 44/62 protein (Fig. 2A), suggesting a lack of holoenzyme complex formation. Furthermore, there is no decrease in the ATPase activity of the 44/62 protein when a 2-fold excess of ΔC6 exo− polymerase (500 nM) is added (data not shown).
Figure 2.
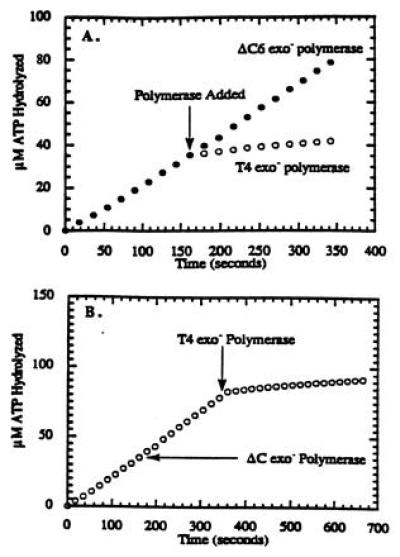
Titration curve for the formation of the bacteriophage T4 holoenzyme complex in which the concentration of 44/62 protein was maintained at 250 nM, while the concentrations of Bio-34/62/36-mer and 45 protein were fixed at 250 nM. Streptavidin was maintained at 1 μM, while the ATP concentration was fixed at 1 mM. Before the addition of either polymerase, the steady-state rate of ATP hydrolysis was 210 nM/s. At the times indicated (arrow), either 250 nM T4 exo− polymerase or 250 nM ΔC6 exo− polymerase was added. (A) The ATPase activity of the 44/62 protein upon the addition of T4 exo− polymerase decreased to eventually reach a limiting rate of 20 nM/s, while the ATPase activity upon the addition of ΔC6 exo− polymerase did not decrease, indicating that the mutant polymerase is incapable of holoenzyme formation. (B) The ATPase activity of the 44/62 protein did not decrease upon the addition of ΔC6 exo− polymerase, but did decrease upon the addition of T4 exo− polymerase, indicating the specific interaction of wild-type polymerase with the 45 protein.
A similar contrasting experiment was performed in which the steady-state rate of ATP hydrolysis using stoichiometric quantities of 44/62 protein, 45 protein, and Bio-34/62/36-mer (in the presence of 1 μM streptavidin) was measured in the absence of either polymerase (Fig. 2B). Upon the addition of 500 nM ΔC6 exo− polymerase, there is no decrease in the rate of ATP hydrolysis by the 44/62 protein (≈220 nM/s). When 250 nM T4 exo− polymerase is added after the ΔC6 exo− polymerase, the ATPase activity of the 44/62 is decreased to 20 nM/s and is indicative of holoenzyme complex formation. Thus, ΔC6 exo− polymerase does not effectively compete with wild-type enzyme for the 45 protein underscoring the importance of the 6 aa at the carboxyl terminus for the protein–protein interaction.
A second method to assess holoenzyme formation involves using the strand displacement assay previously developed by Kaboord and Benkovic (3). Processive DNA synthesis by the holoenzyme complex using the defined Bio-34/62/36-mer DNA substrate (in the presence of streptavidin) results in strand displacement of the 36-mer forked strand to produce fully elongated products (a mixture of 61- and 62-mer). Smaller products (<44-mer) are generated by polymerase not in holoenzyme since polymerase alone cannot perform strand displacement synthesis under the reaction times employed in this study.
Assembly of the holoenzyme complex was performed using either wild-type exo− or ΔC6 exo− polymerase in which a solution of Bio-34/62/36 (500 nM) and streptavidin (1 μM) was incubated with 44/62 protein and 45 protein (550 nM each) in the presence of 1 mM ATP for 10 s. This time frame allows for each DNA molecule to be loaded with a 45 protein. Either 100 nM T4 exo− polymerase or 100 nM ΔCexo− polymerase was then added as well as 10 μM dCTP (the first nucleotide to be incorporated). After ≈5 s, the remaining dNTPs (10 μM each) and single-stranded DNA (to trap free polymerase) were added to initiate DNA synthesis by any assembled holoenzyme complexes. The reaction was manually quenched 10 s after the addition of dNTPs and single-stranded DNA trap by the addition of 1 M HCl.
Production of 62-mer (Fig. 3B) indicates that holoenzyme complex is formed using wild-type exo− polymerase, consistent with previous measurements (3, 4). The ΔC6 exo− polymerase does not form a productive holoenzyme complex, since there are no strand displacement products formed (Fig. 3C) although the ΔC6 exo− polymerase by itself is able to incorporate up to the forked strand (Fig. 3D), as is the case for wild-type exo− polymerase alone (Fig. 3A). Collectively, the lack of holoenzyme assembly by either assay clearly indicates that the carboxyl terminal portion of the polymerase is directly responsible for productive interactions with the 45 protein. Although a crystal structure of the 45 protein has recently been solved (J. Kuriyan, personal communication), there is no obvious structural feature present on the 45 protein to account for its interaction with this 6-aa segment.
Figure 3.
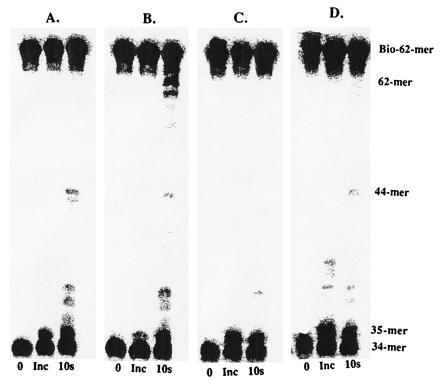
Holoenzyme formation and processive DNA synthesis measurements by wild-type exo− polymerase alone (A), in the presence of accessory proteins (B), or by ΔC6 exo− polymerase alone (C) and in the presence of accessory proteins (D). Assays were performed as described in text.
Effect of Synthetic Peptide on Holoenzyme Complex Formation.
A peptide corresponding to the carboxyl-terminus of the T4 exo− polymerase (Fig. 1) was tested as an inhibitor of holoenzyme formation. Interaction of the peptide with the 45 protein might disrupt the functional interaction of the 45 protein with the polymerase and be manifest in the ATPase activity of the 44/62 protein during the process of clamp loading and ultimately holoenzyme formation. The ATPase activity of the 44/62 protein then would not diminish in the presence of peptide, since the 45 protein will be constantly reloaded onto the DNA (by the ATP-dependent action of the 44/62 protein) and would reflect the inhibition of holoenzyme formation. Thus, by titrating the ATPase activity with variable concentrations of peptide, an inhibition constant (equivalent to KD under these conditions) for the peptide would be obtained reflecting the binding energy of these 6 aa.
However, it was first necessary to verify that the peptide would not “lock” the 45 protein on the DNA by forming a “pseudo-holoenzyme complex” that would also result in a diminished ATPase activity of the 44/62 protein. The ATPase activity of the 44/62 protein in the presence of stoichiometric 45 protein and duplex DNA (20/27-mer) was measured in the presence of peptide A. The rate of ATP hydrolysis by the 44/62 protein decreased linearly as a function of increasing peptide concentrations (0–10 μM), consistent with a simple competitive inhibition model (data not shown). A plot of reciprocal velocities versus peptide concentration is linear, yielding a Ki value for peptide A equal to 1.6 ± 0.2 μM (data not shown). This decrease in ATPase activity is specific for the interaction of the 44/62 protein with the 45 protein, since incremental additions of either 45 protein or 44/62 protein result in linear increases in ATP consumption (data not shown). It is highly unlikely that a pseudo-holoenzyme complex was formed on the DNA that resulted in a diminished ATPase activity of the 44/62 protein, since the DNA substrate used in this experiment was devoid of any physical barriers that would prevent the 45 protein from translocating off the ends of the DNA. The most likely explanation for the decrease in ATPase activity is that peptide A specifically interferes with the 44/62 protein interacting with the 45 protein, ultimately resulting in a lack of 45 protein being loaded onto the DNA. Furthermore, this interference implies that the 44/62 protein and polymerase interact with the same “face” or site of interaction of the 45 protein.
Strand displacement assays were then performed to test if peptide A can inhibit holoenzyme complex formation by disrupting the interaction of the 45 protein with the wild-type exo− polymerase. These assays were performed as previously described except that varying concentrations of peptide A (0–10 μM) were preincubated with the accessory proteins and DNA in the presence of ATP before the addition of wild-type T4 exo− polymerase. As depicted in Fig. 4, holoenzyme complex is formed even at the highest concentration of peptide A (10 μM), as judged by the accumulation of 62-mer product. However, upon close inspection, the gel also reveals smaller elongation products at higher peptide concentrations, indicating that not all of the polymerase is present as the holoenzyme. For example, in the absence of peptide A, there is predominant accumulation of 62-mer product, while very little accumulation of products <44-mer, indicating that all polymerase is present as holoenzyme. However, using 10 μM peptide A, accumulation of products <44-mer indicate free polymerase, while strand displacement products (62-mer) indicate formation of holoenzyme. While it is difficult to accurately quantitate the amount of polymerase not in the holoenzyme complex under these steady-state conditions, the data clearly show a relationship between the concentration of peptide A and inhibition of holoenzyme complex formation.
Figure 4.
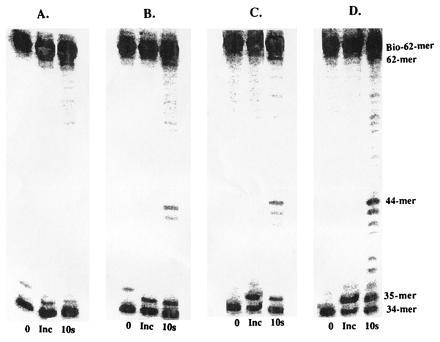
Holoenzyme formation and processive DNA synthesis measurements by wild-type exo− polymerase in the presence of varying concentrations of peptide A. (A) No peptide; (B) 1 μM; (C) 5 μM; and (D) 10 μM. Assays were performed as described in text.
The inhibition of the ATPase activity of the 44/62 protein by peptide A suggests that holoenzyme should not have been formed using the strand displacement assay, especially at the higher peptide concentrations (>5 μM), since the 45 protein is presumably inhibited from being loaded onto the DNA. It is important to note that the ATPase activity of the 44/62 protein only reflects the interaction of the 44/62 protein with the 45 protein during the process of loading the 45 protein onto duplex DNA. While 45 protein is hindered from being loaded onto the DNA in the presence of peptide A, a fraction of the 45 protein will be loaded onto the primer/template under these steady state conditions. Any 45 protein present on the DNA must be able to interact with the polymerase to form the holoenzyme due to the production of strand displacement products. The lack of substantial inhibition of holoenzyme formation under these steady-state conditions suggests that the polymerase–45 protein interaction on DNA is extremely tight. With the koff = 0.003 s−1 (3) and assuming kon = 108 M−1·s−1, we can estimate the equilibrium binding constant of the 45 protein with polymerase on DNA to be at least 30 pM. This low value will reflect a combination of complex interactions involving the polymerase–DNA interaction (KD = 40 nM), the polymerase–45 protein interaction, and the intrinsic interactions of the 45 protein with the DNA.
Conclusion.
DNA replication is a complex process involving numerous protein–nucleic acid interactions as well as protein–protein interactions. We have specifically examined the interaction of the bacteriophage T4 DNA polymerase with its processivity factor, the 45 protein, to address the dynamics and the minimal recognition sequence required for holoenzyme formation. The ΔC6 exo− mutant polymerase is kinetically identical to wild-type exo− polymerase with respect to binding constants for primer/template and nucleotide substrate as well as with respect to pre-steady-state and steady-state rates of nucleotide incorporation. The mutant polymerase is unable to functionally interact with the 45 protein to form the stable holoenzyme required for processive DNA synthesis. Thus, the 6 aa at the carboxyl terminus of the polymerase are absolutely required for interaction(s) with the sliding clamp. A peptide designed to mimic the carboxyl terminus of the polymerase inhibits the interaction of the 44/62 protein with the 45 protein. Inhibiting the functional interactions of the 44/62 protein with the 45 protein by the peptide designed to mimic the polymerase implies that the 44/62 protein and polymerase interact with the 45 protein at a common site. The shared site of interaction may have implications with regards to the chaperone function of the 44/62 protein during holoenzyme assembly (24).
The use of peptidomimetics as therapeutic agents has recently been employed against herpes simplex virus replication in vitro (7). Similar to our results, the carboxyl-terminus of the herpes simplex virus polymerase interacts specifically with the UL42 accessory protein (the functional equivalent of the bacteriophage T4 45 protein) to increase the polymerase’s low degree of processivity (10). Large peptides corresponding to the carboxyl terminus of the polymerase have been used to disrupt the interaction of the polymerase with UL42, thus effectively inhibiting processive DNA synthesis by the herpes simplex holoenzyme in vitro. However, a high concentration of peptide (100 μM) was used to inhibit all processive DNA synthesis, making its effectiveness in vivo speculative. Nevertheless, that small peptide fragments can specifically disrupt the interactions between two large proteins involved in an essential and complex biological process such as DNA replication is encouraging for further developments of peptides as therapeutic agents.
One major complication in disrupting the protein–protein interactions involved in DNA replication is that the peptide used in our studies most likely does not bind to DNA. Thus, the peptide can only intercept the accessory proteins before binding to DNA. As a result, there is very little chance of disrupting the interaction of the polymerase with its processivity factor once the functional holoenzyme is assembled on DNA. A more effective approach to inhibiting biological processes that utilize nucleic acids would be to prevent the association of the two proteins before complex formation by linking the peptidomimetic to another molecule (possibly another protein) that is also capable of tightly binding nucleic acid. This method would first provide a more structurally related analog of the protein complex to be inhibited (protein–DNA complex). Secondly, this approach would also improve inhibition by removing kinetic barriers imposed by a three-dimensional search (peptide free in solution) as opposed to a two-dimensional search (inhibition complex translocating/bound to DNA).
Acknowledgments
This work supported in part by a National Institutes of Health Fellowship GM 16704 (A.J.B.) and a U.S. Public Health Service Grant GM 13306 (S.J.B.). We deeply thank Dr. Michael Moore of SmithKline Beecham for the generous gift of the polypeptides used in this study.
References
- 1.Nossal N G. FASEB J. 1992;6:871–878. doi: 10.1096/fasebj.6.3.1310946. [DOI] [PubMed] [Google Scholar]
- 2.Young M C, Reddy M K, von Hippel P H. Biochemistry. 1992;31:8675–8690. doi: 10.1021/bi00152a001. [DOI] [PubMed] [Google Scholar]
- 3.Kaboord B F, Benkovic S J. Curr Biol. 1995;5:149–157. doi: 10.1016/s0960-9822(95)00036-4. [DOI] [PubMed] [Google Scholar]
- 4.Berdis A J, Benkovic S J. Biochemistry. 1996;35:9253–9265. doi: 10.1021/bi952569w. [DOI] [PubMed] [Google Scholar]
- 5.Krishna T S R, Kong X-P, Gary S, Burgers P M, Kuriyan J. Cell. 1994;79:1233–1243. doi: 10.1016/0092-8674(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 6.Kong X-P, Onrust R, O’Donnell M, Kuriyan J. Cell. 1992;69:425–437. doi: 10.1016/0092-8674(92)90445-i. [DOI] [PubMed] [Google Scholar]
- 7.Digard P, Williams K P, Hensley P, Brooks I S, Dahl C E, Coen D M. Proc Natl Acad Sci USA. 1995;92:1456–1460. doi: 10.1073/pnas.92.5.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaboord B F, Benkovic S J. Proc Natl Acad Sci USA. 1993;90:10881–10885. doi: 10.1073/pnas.90.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hernandez T R, Lehman I R. J Biol Chem. 1990;265:11227–11232. [PubMed] [Google Scholar]
- 10.Digard P, Bebrin W, Weisshart K, Coen D M. J Virol. 1993;67:1159–1168. doi: 10.1128/jvi.67.1.398-406.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stow N. Nucleic Acids Res. 1993;21:87–92. doi: 10.1093/nar/21.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berthomme H, Monahan S J, Parris D S, Jacquemont B, Epstein A L. J Virol. 1995;69:2811–2818. doi: 10.1128/jvi.69.5.2811-2818.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Venkatesan M, Nossal N G. J Biol Chem. 1982;257:12435–12443. [PubMed] [Google Scholar]
- 14.Herendeen D R, Kassavetis G A, Barry J, Alberts B M, Geidushek E P. Science. 1989;245:952–958. doi: 10.1126/science.2672335. [DOI] [PubMed] [Google Scholar]
- 15.Tinker R L, Sanders G M, Severinov K, Kassavetis G A, Geidushek E P. J Biol Chem. 1995;270:15899–15907. doi: 10.1074/jbc.270.26.15899. [DOI] [PubMed] [Google Scholar]
- 16.Winkelman J W, Kassavetis G A, Geidushek E P. J Bacteriol. 1994;176:1164–1171. doi: 10.1128/jb.176.4.1164-1171.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Capson T L, Peliska J A, Kaboord B F, Frey M W, Lively C, Dahlberg M, Benkovic S J. Biochemistry. 1992;31:10984–10994. doi: 10.1021/bi00160a007. [DOI] [PubMed] [Google Scholar]
- 18.Kuchta R D, Mizrahi V, Benkovic P A, Johnson K A, Benkovic S J. Biochemistry. 1987;26:8410–8417. doi: 10.1021/bi00399a057. [DOI] [PubMed] [Google Scholar]
- 19.Frey M W, Nossal N G, Capson T L, Benkovic S J. Proc Natl Acad Sci USA. 1993;90:2579–2583. doi: 10.1073/pnas.90.7.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jarvis T C, Paul L S, von Hippel P H. J Biol Chem. 1989;264:12709–12716. [PubMed] [Google Scholar]
- 21.Johnson K A. Methods Enzymol. 1986;134:677–705. doi: 10.1016/0076-6879(86)34129-6. [DOI] [PubMed] [Google Scholar]
- 22.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1982. [Google Scholar]
- 23.Mizrahi V, Benkovic P A, Benkovic S J. Proc Natl Acad Sci USA. 1986;83:5769–5773. doi: 10.1073/pnas.83.16.5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaboord B F, Benkovic S J. Biochemistry. 1996;35:1084–1092. doi: 10.1021/bi9520747. [DOI] [PubMed] [Google Scholar]