

Abstract
In one form of β-thalassemia, a genetic blood disorder, a mutation in intron 2 of the β-globin gene (IVS2-654) causes aberrant splicing of β-globin pre-mRNA and, consequently, β-globin deficiency. Treatment of mammalian cells stably expressing the IVS2-654 human β-globin gene with antisense oligonucleotides targeted at the aberrant splice sites restored correct splicing in a dose-dependent fashion, generating correct human β-globin mRNA and polypeptide. Both products persisted for up to 72 hr posttreatment. The oligonucleotides modified splicing by a true antisense mechanism without overt unspecific effects on cell growth and splicing of other pre-mRNAs. This novel approach in which antisense oligonucleotides are used to restore rather than to down-regulate the activity of the target gene is applicable to other splicing mutants and is of potential clinical interest.
Keywords: RNA splicing, gene therapy
β-Thalassemia, a genetic blood disorder, affects a large number of people in the Mediterranean basin, Middle East, South East Asia, and Africa. Close to 100 thalassemic mutations causing defective β-globin gene expression and β-globin deficiency have been identified, but no more than 10 mutations are responsible for ≈90% of cases worldwide (1). Of the frequently occurring mutations, the ones that cause aberrant splicing of intron 1 of the human β-globin gene are predominant in South Eastern Europe, Cyprus, Lebanon (mutation IVS1-110), India, Malaysia, and Indonesia (IVS1-5). Additional splicing mutations in intron 1 (IVS1-6) as well as in intron 2 of the β-globin gene (IVS2-745) are also common in the above countries, while IVS2-654 is frequent among β-thalassemia patients in China and Thailand (1–8). All of these mutations activate aberrant splice sites and change the splicing pathway even though the correct splice sites remain potentially functional. We hypothesized that blocking of the aberrant splice sites or other sequence elements involved in splicing with antisense oligonucleotides may force the splicing machinery to reselect the correct splice sites and induce the formation of β-globin mRNA and polypeptide, hence restoring the gene function.
Although we have previously effected correction of splicing by antisense oligonucleotides in cell-free extracts from HeLa cells (9), it was not at all clear whether the oligonucleotides delivered into the cell could enter the nucleus, hybridize to the aberrant splice sites in competition with the splicing factors, and promote the formation of the spliceosome and subsequent splicing at the correct splice site. Here we report that correct splicing was efficiently restored when phosphorothioate 2′-O-methyl-oligoribonucleotides were targeted to the aberrant splice sites of IVS2-654 pre-mRNA expressed in mammalian cells stably transformed with this mutated human β-globin gene. This is a novel approach since antisense oligonucleotides have been used mostly as sequence specific down-regulators of gene expression (10).
MATERIALS AND METHODS
Cells.
Human β-globin gene carrying a thalassemic mutation IVS2-654 was cloned under the cytomegalovirus promoter (11). The plasmid was cotransfected with a neomycin resistance plasmid by lipofection with Lipofectamine (GIBCO/BRL) into HeLa and NIH 3T3 cells, and the cells stably expressing the IVS2-654 β-globin gene were isolated by G-418 antibiotic selection. Control cells expressing the wild-type gene were obtained in a similar manner. HeLa and NIH 3T3 cell lines were grown in MEM supplemented with 5% fetal calf and 5% horse sera and in DMEM, high glucose, with 10% filtered Colorado calf serum, respectively. For all experiments, cells were plated in 24-well plates at 105 cells per well 24 hr before treatment.
Oligonucleotide Treatment.
The phosphorothioate 2′-O-methyl-oligoribonucleotides (prepared and purified at Hybridon) were used. The cells were treated with oligonucleotides complexed with Lipofectamine for 10 and 6 hr for HeLa and NIH 3T3 cell lines, respectively (12, 13). In Figs. 1 B and C, 3A, and 4B, the cells were harvested 36 hr later and were subsequently analyzed. The oligonucleotides 5′ss-GCUAUUACCUUAACCCAG and 3′ss-CAUUAUUGCCCUGAAAG were targeted to the aberrant 5′ splice site and the 3′ cryptic splice site, respectively. Oligonucleotides with random or scrambled sequences were used as controls. An oligonucleotide, CCUCUUACCUCAGUUACA, targeted to positions 696–713 of β-globin intron 2, encompassing thalassemic mutation IVS2-705 (8), was used as an additional control.
Figure 1.
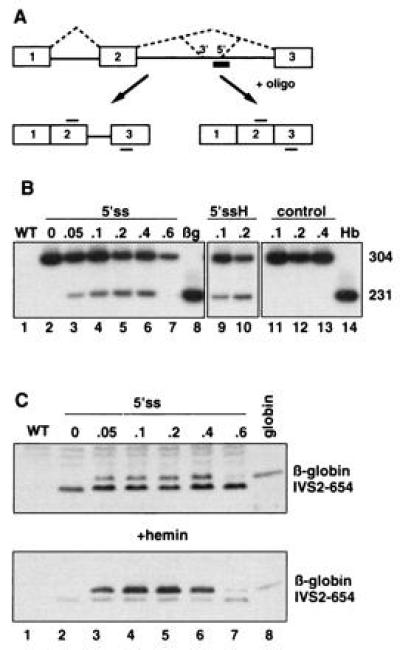
(A) Splicing of human β-globin IVS2-654 pre-mRNA in the presence of an antisense oligonucleotide. Boxes, exons; solid lines, introns; dashed lines, both correct and aberrant splicing pathways; thick bar, oligonucleotide antisense to the aberrant 5′ splice site; thin bars above and below exon sequences, primers used in the RT-PCR reaction. The aberrant 5′ splice site created by IVS2-654 mutation and the cryptic 3′ splice site activated upstream are indicated. (B) Correction of splicing of IVS2-654 pre-mRNA in HeLa cells by antisense oligonucleotide targeted to the aberrant 5′ splice site (5′ss). Analysis of total RNA by RT-PCR. Lanes: 1, wild-type (WT) HeLa cells; 8, HeLa cell line expressing normal human β-globin (βg); 14, RNA from human blood (Hb); 2–7, IVS2-654 HeLa cells treated with increasing concentrations of the oligonucleotide (indicated in micromoles at the top); 9 and 10, IVS2-654 HeLa cells treated with oligonucleotide followed by hemin (H) (16); 11–13, IVS2-654 HeLa cells treated with increasing concentrations of the scrambled oligonucleotide. The numbers on the right indicate the size, in nucleotides, of the RT-PCR products representing the aberrantly (304) and correctly (231) spliced RNAs. (C) Restoration of β-globin expression by 5′ss oligonucleotide in IVS2-654 HeLa cells. Immunoblot of total protein with anti-human hemoglobin antibody. Concentration of the oligonucleotide in micromoles is indicated at the top (lanes 2–7); in lane 8, human globin (Sigma) was used as a marker. (Lower) Cells were treated with hemin preceding the isolation of proteins. The positions of human β-globin and the prematurely terminated β-globin IVS2-654 polypeptide are indicated. Time of exposure of the autoradiogram in Lower was 1/5th of that of the Upper.
Figure 3.
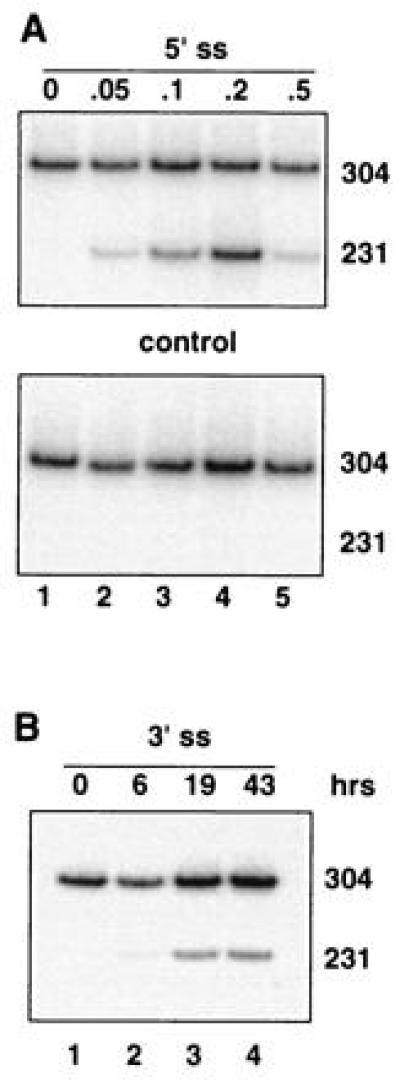
Dose- and time-dependent correction of splicing in oligonucleotide-treated IVS2-654 NIH 3T3 cells. RT-PCR assay. (A) Cells treated with increasing concentrations of the oligonucleotide targeted to the aberrant 5′ splice site (Upper) or of the control, scrambled oligonucleotide (Lower). (B) Time course of the correction of splicing after termination of treatment with 0.2 μM oligonucleotide targeted to the cryptic 3′ splice site activated by the IVS2-654 mutation. All designations are as in Figs. 1 and 2.
RNA Analysis.
Total RNA was isolated with TRI-Reagent (Molecular Research Center, Cincinnati) and analyzed by reverse transcription–PCR (RT-PCR) using rTth DNA polymerase as suggested by the manufacturer (Perkin–Elmer). Forward and reverse primers spanned positions 21–43 of exon 2 of the human β-globin gene and positions 6–28 of exon 3, respectively. The RT-PCR products were separated on 7.5% nondenaturing polyacrylamide gel. To ascertain that the protocol is suitable for quantitative analysis, the RT-PCR was carried out with [α-32P]dATP for no more than 18–20 cycles. Under these conditions, the amount of the PCR product is proportional to the amount of input RNA as are the relative amounts of PCR products generated from aberrantly and correctly spliced RNA (ref. 14 and data not shown). No product is detectable without the reverse transcription step.
Protein Analysis.
Hemin (10 μM, Fluka) treatment was in serum-free medium for 4 hr immediately preceding the isolation of RNA or protein. Blots of proteins separated on a Tricine-SDS/10% polyacrylamide gel (15) were incubated with polyclonal affinity-purified chicken anti-human hemoglobin IgG as primary antibody and rabbit anti-chicken horseradish peroxidase-conjugated IgG as secondary antibody (Accurate Chemicals). Subsequently, the blots were developed with the Enhanced Chemiluminescence detection system (Amersham).
All autoradiograms were captured by a DAGE–MTI (Michigan City, IN) CCD72 video camera, and the images were processed using National Institutes of Health image 1.47 and macdraw pro 1.0 software. The image 1.47 was also used for quantitation of the autoradiograms. The final figures were printed on Tektronix phaser 440 printer.
RESULTS
Since appropriate cellular or animal models of thalassemic splice mutants are not available, we have constructed two cell lines stably transformed with the IVS2-654 variant of the thalassemic human β-globin gene. In a HeLa-based cell line, as in thalassemic patients (1, 3, 4), this mutation created a 5′ splice site at nucleotide 652 of intron 2 and activated a 3′ cryptic splice site 73 nucleotides upstream, resulting in stably expressed but aberrantly spliced IVS2-654 β-globin pre-mRNA (Fig. 1 A and B, lane 2). To restore correct splicing of the RNA, the cells were treated for 10 hr with a complex of Lipofectamine and the 18-mer phosphorothioate 2′-O-methyl-oligoribonucleotide (5′ss) targeted to the aberrant 5′ splice site. The 2′-O-methyl derivatives were chosen since they hybridize well to their target sequences and are very stable in cellular environment. Moreover, importantly, in contrast to commonly used oligodeoxynucleotides or phosphorothioate oligodeoxynucleotides, they do not promote cleavage of hybridized RNA by cellular RNase H (17). The latter property is the key condition for the success of the experiments since treatment with an unmethylated oligonucleotide would have led to degradation of the β-globin pre-mRNA and removal of the splicing substrate (10, 18).
Fig. 1B shows that treatment with 2′-O-methyl phosphorothioates was effective in blocking the aberrant splice site and restoring correct splicing of β-globin pre-mRNA. Quantitative RT-PCR analysis (ref. 14; see Materials and Methods) of the RNA showed that the amount of correctly spliced β-globin mRNA increased in a dose-dependent fashion, and at 0.05, 0.1, and 0.2 μM oligonucleotide reached, respectively, 16, 24, and 34% of the total (Fig. 1B, lanes 3–5 and Table 1). There was no further increase in the correctly spliced product at 0.4 μM oligonucleotide (33%), while treatment at 0.6 μM oligonucleotide drastically lowered its amount (Fig. 1B, lanes 6 and 7, respectively). The latter result is possibly due to the fact that the ratio of Lipofectamine:nucleic acid deviated from a narrow range necessary for efficient cellular uptake of the complex (13). The effect of the antisense oligonucleotide was sequence-dependent since control oligonucleotides either with random or with scrambled sequences (Fig. 1B, lanes 11–13) did not restore correct splicing. Somewhat weaker correction of aberrant splicing of IVS2-654 pre-mRNA (11%) was obtained when the cells were treated with a 17-mer oligonucleotide antisense to the 3′ cryptic splice site activated by the IVS2-654 mutation (see Table 1). Note that in untreated (Fig. 1B, lane 2) or control (Fig. 1B, lanes 11–13) cells, there was no detectable PCR product representing the correctly spliced β-globin mRNA. Therefore, in both 5′ss and 3′ss oligonucleotide-treated cells the β-globin mRNA must have been spliced de novo and the observed band could not have resulted from preferential RT-PCR amplification of a preexisting shorter, correctly spliced mRNA.
Table 1.
Quantitation of correct expression of β-globin mRNA and protein
| Target cell line and splice site | % correct β-globin product |
|---|---|
| HeLa 5′ss* | 34 |
| HeLa 5′ss† | 14 |
| HeLa 5′ss* | 43 (protein) |
| HeLa 3′ss* | 11 |
| NIH 3T3 5′ss† | 49 |
| NIH 3T3 3′ss† | 23 |
The results of the treatment with 0.2 μM antisense oligonucleotides, the concentration that elicits maximal correction in all experiments, are shown. The amount of the material in the correct PCR product or in β-globin protein band was quantitated by densitometry of the autoradiograms as described. The results are expressed as percent of the correct product relative to the sum of correct and aberrant products.
Treatment with oligonucleotide was for 10 hr.
Treatment with oligonucleotide was for 6 hr.
Analysis of the total protein from oligonucleotide-treated cells by immunoblotting with polyclonal antibody to human hemoglobin showed that the newly generated, correctly spliced β-globin mRNA was translated into full-length β-globin. In agreement with the RT-PCR results shown in Fig. 1B, only samples treated with 0.05–0.4 μM oligonucleotide contained significant amounts of full-length β-globin (Fig. 1C, lanes 3–6). There was no β-globin in control cells (Fig. 1C, lanes 1 and 2) and only a small amount in those treated with 0.6 μM oligonucleotide (Fig. 1C, lane 7). Thus, the significant increase in full-length β-globin, roughly parallel to that of the β-globin mRNA, is clearly due to the effect of antisense oligonucleotides on splicing. The quantitative analysis of the amount of the β-globin polypeptide relative to the one truncated due to aberrant splicing (in the aberrant sequence the stop codon is located 48 nucleotides downstream from exon 2, resulting in a β-globin polypeptide containing 104 β-globin and 16 aberrant amino acids) shows that the amount of β-globin increases from ≈30% of the total at 0.05 μM oligonucleotide to 43% at 0.2 μM and 44% at 0.4 μM oligonucleotide. The fact that the percentage of β-globin protein seems to be slightly higher than that of the corresponding correctly spliced mRNA may possibly be due to the differences in the relative stabilities of the correct and aberrant polypeptides. Nevertheless, the yields of correct protein provide evidence that the amount of the correctly spliced β-globin mRNA is not overrepresented in the RT-PCR assay.
The identity of the generated full-length β-globin polypeptide band was confirmed by the increase in its intensity upon posttreatment of the cells with hemin (Fig. 1C Lower, lanes 3–6) (16). Note that hemin treatment had no effect on the truncated IVS2-654 polypeptide or background protein bands (Fig. 1C Upper and Lower, lanes 2–7). Neither did it affect the level of transcription and splicing pattern of the IVS2-654 pre-mRNA (Fig. 1B, lanes 9 and 10). Thus, the increase in β-globin band due to hemin is not the result of activation of globin gene expression, observed for fetal globin genes in hematopoietic cell lines (e.g., ref. 19 and references therein). It seems likely that the polyclonal anti-hemoglobin antibody has greater affinity for the β-globin-heme complex than for β-globin alone and/or that hemin treatment results in specific posttranslational stabilization of the full-length β-globin (20).
Fig. 2 shows the time course of restoration of correct splicing of β-globin pre-mRNA and its translation to protein after treatment with 0.2 μM 5’ss oligonucleotide. Six hours after termination of the treatment, there was a trace, if any, of the correct β-globin mRNA and protein (Fig. 2 A, lane 3, and B, lane 2, respectively) that increased significantly at 24 hr and persisted for 48 but not 96 hr (Fig. 2 A, lanes 4–6, and B, lanes 3–5). The β-globin signal was, as expected, stimulated by hemin treatment of the cells (Fig. 2B, lane 6 versus lane 3). The fact that correctly spliced RNA persisted for 48 hr after termination of oligonucleotide delivery suggests that the oligonucleotides and/or the newly synthesized correctly spliced mRNA are quite stable in the cellular environment. It is also possible that the oligonucleotide is recycled after the spliced out intron is degraded (21).
Figure 2.
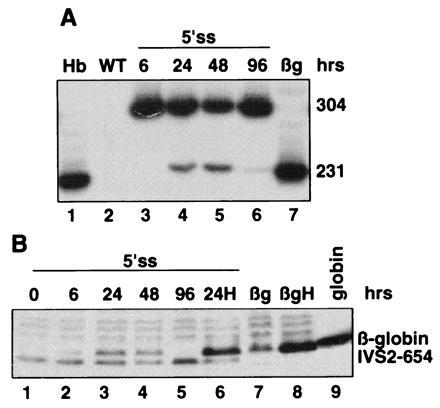
Time course of restoration of correct splicing and β-globin expression in HeLa IVS2-654 cells by 0.2 μM 5′ss oligonucleotide. (A) RT-PCR assay. (B) Immunoblot. Time after termination of oligonucleotide treatment is indicated at the top. H, hemin treatment of the cells. All other designations are as in Fig. 1.
To test whether oligonucleotides are able to reverse aberrant splicing in other cell types, analogous experiments were performed using NIH 3T3 cells stably transfected with the IVS2-654 β-globin gene. Since 10 hr incubation in the serum-free medium (used for HeLa cells) was damaging for the NIH 3T3 cells, the treatment was shortened to 6 hr. Even with the shortened treatment, 5′ss oligonucleotide targeted to the aberrant 5′ splice site in IVS2-654 pre-mRNA produced correctly spliced β-globin mRNA at levels ≈3-fold higher than those observed for HeLa cells treated with the same oligonucleotide for the same time (Table 1). As expected, the effects of the oligonucleotide were dose- and sequence-dependent (Fig. 3A).
Repair of aberrant splicing was also obtained, albeit not as efficiently, by targeting the 3′ cryptic splice site (Fig. 3B and Table 1). This indicates that the relative accessibility of the 3′ versus 5′ splice site is similar in both HeLa and NIH 3T3 cells. The time course of the reaction (Fig. 3B) suggests that there is no major difference in the stability of the β-globin mRNA and of the two oligonucleotides in the two cell types.
Although the above results clearly show that the oligonucleotides affect splicing of their target pre-mRNAs in a sequence specific manner, one cannot exclude the possibility that they may exert other effects on the cells. The oligonucleotides may interact directly with cellular proteins (ref. 22 and references therein) or, possibly, inhibit gene expression by blocking similar splice sites in many other pre-mRNAs and consequently inhibit the growth of cells. However, the results presented in Fig. 4 show that under our experimental conditions no unspecific effects were detectable. First, the growth rate of the HeLa IVS2-654 cells treated with the Lipofectamine–oligonucleotide complex was no different from that of cells treated with Lipofectamine alone (Fig. 4A). The oligonucleotides tested included 2′-O-methyl phosphorothioates complementary to the aberrant splice sites or with a scrambled sequence as well as 5′ss and 3′ss 2′-O-methyl phosphodiesters. Second, the 5′ ss oligonucleotide that restored correct splicing in HeLa IVS2-654 (Fig. 4B, lane 6) had no effect on splicing of HeLa cells transfected with a control construct in which the target aberrant 5′ splice site (GUAAUA) was modified to match the consensus splice site sequence (GUAAGU; ref. 23) (Fig. 4B, lanes 2–4). This modification resulted in a two nucleotide mismatch of the oligonucleotide with 16 nucleotides remaining complementary to the intron sequence. Third, splicing of IVS2-654 pre-mRNA was not affected in cells treated with an oligonucleotide with partial complementarity to the region of the aberrant 5′ splice site (slash indicates splice site):
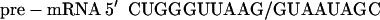 |
 |
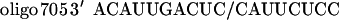 |
Figure 4.
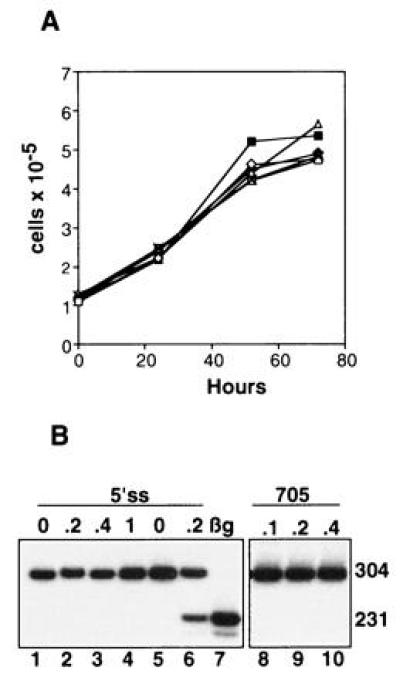
Specificity of oligonucleotide treatments. (A) Lack of effect of oligonucleotides on cell growth. HeLa IVS2-654 cells were treated with Lipofectamine–oligonucleotide complexes as described. Cells were counted at the end of the 10 hr treatment (0 hr) and at 24, 52, and 72 hr thereafter. Each point on the curve represents the average of duplicate counts of two independently treated samples; the observed differences are within experimental error (one SD). □, Lipofectamine alone. The remaining samples were treated with Lipofectamine complexed with the following 0.2 μM oligoribonucleotides: 2′-O-methyl phosphorothioates, ⋄, 5′ss, ×, 3′ss, ♦, scrambled; 2′-O-methyl phosphodiesters, ▵, 5′ss, ▪, 3′ss. (B) Lack of effect of control oligonucleotides. Treatment of HeLa IVS2-654 consensus cell line (lanes 1–4, see text) or HeLa IVS2-654 cell line (lanes 5 and 6, as positive control) with 5′ss 2′-O-methyl phosphorothioate oligoribonucleotide. Lanes 8–10, treatment of HeLa IVS2-654 cells with oligonucleotide 705 targeted 44 nucleotides downstream from the aberrant 5′ splice site (see text). The RT-PCR assay and all designations are as described in the legend to Fig. 1B. Lane 7, HeLa cell line expressing normal human β-globin.
Moreover, this oligonucleotide, designed to restore correct splicing of IVS2-705 thalassemic mutant (ref. 8; unpublished work) is also complementary, with a single mismatch, to positions 696–713 of IVS2, 44 nucleotides downstream from the aberrant 5′ splice site (Fig. 4B, lanes 8–10).
Since the closely related IVS2-654 consensus splice site is unaffected by the 5′ss oligonucleotide, and a related oligonucleotide, oligo 705, with partial complementarity to two sites in the same RNA has no effect on splicing of IVS2-654 pre-mRNA, the likelihood that the 5′ss oligonucleotide would affect splicing of other pre-mRNAs with even more divergent sequences at and around the splice sites appears quite remote. This is further reinforced by the fact that we have not detected any changes in the level and/or the splicing patterns of two randomly chosen mRNAs (β-actin and EGFR) in cells treated with the 5′ss oligonucleotide (not shown) and by the data from GenBank that show lack of complementarity of any human sequence besides β-globin to the 5′ss and 3′ss oligonucleotides, even if two mismatches are allowed.
DISCUSSION
We showed that splicing pathways can be modified in vivo in a sequence specific manner by antisense oligonucleotides using cationic liposomes as a carrier. In view of the universal nature of the splicing mechanisms, this approach is of general applicability because the oligonucleotides should be effective in different cell types, including hematopoietic cells, and against splice sites in a variety of pre-mRNAs.
Although the feasibility of treatment of thalassemics with antisense oligonucleotides has yet to be explored, several observations suggest that this approach may be clinically promising. The optimal effect of oligonucleotides was seen at 0.2–0.4 μM, a concentration achieved in bone marrow of experimental animals (24). Restoration in a patient of β-globin mRNA to 20–30% of the normal level would be of therapeutic significance because heterozygotes with 50% of hemoglobin are frequently asymptomatic while the status of patients undergoing transfusion therapy, with even lower hemoglobin levels, is markedly improved. Furthermore, β-globin mRNA and protein are very stable and so are mature erythrocytes, with a lifespan of about 120 days (1). Thus, in principle, treatment with antisense oligonucleotides may have an extended effect on the in vivo levels of β-globin mRNA and blood hemoglobin, reducing the need for frequent administration. In this context, it is encouraging that the correctly spliced β-globin mRNA and protein generated by a single delivery of the antisense oligonucleotide persisted in NIH 3T3 and HeLa cells for up to 48 hr. Moreover, the fact that it was possible to effectively deliver the oligonucleotides to the nuclei of various cell types suggests that it should be feasible to find appropriate conditions and/or carriers for delivery of the oligonucleotides into cells of patients, including the targeted nucleated erythroblasts. The effects of antisense oligonucleotides should be highly specific because only the latter cells contain the target sequence.
The restoration of correct splicing by targeting the cryptic 3′ splice site (Fig. 3B) is of particular interest since this splice site is activated in other β-thalassemia mutations besides IVS2-654, i.e., IVS2-745 and IVS2-705 (1, 8). Thus, a single oligonucleotide should be effective in correcting splicing in all three mutants, which in clinical setting would translate into a larger number of patients. It is also likely that aberrant splice sites activated by other thalassemic mutations (1, 2) will be amenable to this approach. Since splicing mutations are responsible for a large proportion of β-thalassemia patients (2), it appears that restoration of correct splicing of β-globin pre-mRNA may offer a useful alternative to current treatments (1) and to potential therapies based on stimulation of fetal hemoglobin production (25, 26) or β-globin gene transfer.
The specificity of phosphorothioate antisense oligonucleotides and their mechanism of action were recently questioned (22). However, several lines of evidence indicate that in this work correction of splicing occurred by a true antisense mechanism. The effects were sequence specific because only the oligonucleotides targeted to the splice sites but not the control ones (random and scrambled) restored correct splicing. The oligonucleotide complementary to the aberrant 5′ splice site in IVS2-654 pre-mRNA had no corrective effect on splicing in HeLa cells of a modified, control IVS2-654 consensus construct. There was likewise no corrective effect of an oligonucleotide (oligo 705) that can hybridize to another site of the intron, indicating that the region around the splice sites is essential as a specific target. The same oligonucleotide shows that hybridization with less than 17 consecutive nucleotides gives no antisense effect, indicating that it is unlikely that splicing of unrelated pre-mRNAs may be affected to a significant degree by oligonucleotides targeting IVS2-654. The lack of multiple unspecific effects is also supported by the observation that none of the tested oligonucleotides affected the growth rate of the treated cells. Correction of splicing was observed with 2′-O-methyl-oligoribonucleotides both with and without the phosphorothioate modification (not shown) in IVS2-654 HeLa and NIH 3T3 cells, i.e., regardless of cell type and species. Hence, it seems highly unlikely that the effects of phosphorothioate oligonucleotides were caused by their direct interaction with a cellular protein as observed in several other investigations (ref. 22 and references therein).
Acknowledgments
We thank Elizabeth Smith for technical assistance. This work was supported by a National Institutes of Health grant to R.K.
Footnotes
Abbreviation: RT-PCR, reverse transcription–PCR.
References
- 1.Schwartz E, Benz E J. In: Hematology, Basic Principles and Practice. Hoffman R, Benz E J, Shattil S J, Furie B, Cohen H J, editors. New York: Churchill Livingstone; 1995. pp. 586–610. [Google Scholar]
- 2.Huisman T H J. Br J Haematol. 1990;75:454–457. doi: 10.1111/j.1365-2141.1990.tb07781.x. [DOI] [PubMed] [Google Scholar]
- 3.Cheng T-C, Orkin S H, Antonorakis S E, Potter M J, Sexton J P, Giardina P J V, Li A, Kazazian H H. Proc Natl Acad Sci USA. 1984;81:2821–2826. doi: 10.1073/pnas.81.9.2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang S-Z, Zeng F-Y, Ren Z-R, Lu Z-H, Rodgers G P, Schechter A N, Zeng Y-T. Br J Haematol. 1994;88:541–546. doi: 10.1111/j.1365-2141.1994.tb05071.x. [DOI] [PubMed] [Google Scholar]
- 5.Busslinger M N, Moschonas N, Flavell R A. Cell. 1981;27:289–296. doi: 10.1016/0092-8674(81)90412-8. [DOI] [PubMed] [Google Scholar]
- 6.Fukumaki Y, Ghosh P K, Benz E J, Reddy V B, Lebovitz P, Forget B G, Weissman S M. Cell. 1982;28:585–594. doi: 10.1016/0092-8674(82)90213-6. [DOI] [PubMed] [Google Scholar]
- 7.Treisman R, Orkin S H, Maniatis T. Nature (London) 1983;302:591–595. doi: 10.1038/302591a0. [DOI] [PubMed] [Google Scholar]
- 8.Dobkin C, Bank A. J Biol Chem. 1985;260:16332–16337. [PubMed] [Google Scholar]
- 9.Dominski Z, Kole R. Proc Natl Acad Sci USA. 1993;90:8673–8677. doi: 10.1073/pnas.90.18.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crooke S T, Lebleu B, editors. Antisense Research and Applications. Boca Raton, FL: CRC; 1993. [Google Scholar]
- 11.Dominski Z, Kole R. Mol Cell Biol. 1991;11:6075–6083. doi: 10.1128/mcb.11.12.6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bennett C F, Chiang M Y, Chan H, Shoemaker J, Mirabelli C K. Mol Pharmacol. 1992;41:1023–1033. [PubMed] [Google Scholar]
- 13.Hawley-Nelson P, Ciccarone V, Gebeyehu G, Jessee J, Felgner P L. Focus (Rochester, NY) 1993;15:73–79. [Google Scholar]
- 14.Chen I T, Chasin L A. Mol Cell Biol. 1993;13:289–300. doi: 10.1128/mcb.13.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schagger H, von Jagov G. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 16.London I M, Clemens M J, Ranu R S, Levin D H, Cherbas L F, Ernst V. Fed Proc Fed Am Soc Exp Biol. 1976;35:2218–2222. [PubMed] [Google Scholar]
- 17.Sproat B S, Lamond A I. In: Antisense Research and Applications. Crooke S T, Lebleu B, editors. Boca Raton, FL: CRC; 1993. pp. 351–363. [Google Scholar]
- 18.Furdon P F, Dominski Z, Kole R. Nucleic Acids Res. 1989;17:9193–9204. doi: 10.1093/nar/17.22.9193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fibach E, Kollia P, Schechter A N, Noguchi C T, Rodgers G P. Blood. 1995;85:2967–2974. [PubMed] [Google Scholar]
- 20.Weatherall D J. In: The Molecular Basis of Blood Diseases. Stamatoyannopoulos G, Neinhuis A W, Leder P, Majerus P W, editors. Philadelphia: Saunders; 1994. pp. 157–205. [Google Scholar]
- 21.Green M R. Annu Rev Cell Biol. 1991;7:559–599. doi: 10.1146/annurev.cb.07.110191.003015. [DOI] [PubMed] [Google Scholar]
- 22.Stein C A. Nat Med. 1995;1:1119–1121. doi: 10.1038/nm1195-1119. [DOI] [PubMed] [Google Scholar]
- 23.Dominski Z, Kole R. Mol Cell Biol. 1994;14:7445–7454. doi: 10.1128/mcb.14.11.7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang R, Lu Z, Zhao H, Zhang X, Diasio R B, Habus I, Jiang Z, Iyer R P, Yu D, Agrawal S. Biochem Pharmacol. 1995;50:545–556. doi: 10.1016/0006-2952(95)00159-w. [DOI] [PubMed] [Google Scholar]
- 25.Sher G D, Ginder G D, Little J, Yang S, Dover G J, Olivieri N F. N Engl J Med. 1995;332:1606–1610. doi: 10.1056/NEJM199506153322404. [DOI] [PubMed] [Google Scholar]
- 26.Voskaridou E, Kalotychou V, Loukopoulos D. Br J Haematol. 1995;89:479–484. doi: 10.1111/j.1365-2141.1995.tb08352.x. [DOI] [PubMed] [Google Scholar]