

Abstract
Cell-cycle progression is mediated by a coordinated interaction between cyclin-dependent kinases and their target proteins including the pRB and E2F/DP-1 complexes. Immunoneutralization and antisense experiments have established that the abundance of cyclin D1, a regulatory subunit of the cyclin-dependent kinases, may be rate-limiting for G1 phase progression of the cell cycle. Simian virus 40 (SV40) small tumor (t) antigen is capable of promoting G1 phase progression and augments substantially the efficiency of SV40 transformation through several distinct domains. In these studies, small t antigen stimulated cyclin D1 promoter activity 7-fold, primarily through an AP-1 binding site at −954 with additional contributions from a CRE site at −57. The cyclin D1 AP-1 and CRE sites were sufficient for activation by small t antigen when linked to an heterologous promoter. Point mutations of small t antigen between residues 97–103 that reduced PP2A binding were partially defective in the induction of the cyclin D1 promoter. These mutations also reduced activation of MEK1 and two distinct members of the mitogen-activated protein kinase family, the ERKs (extracellular signal regulated kinases) and the SAPKs (stress-activated protein kinases), in transfected cells. Dominant negative mutants of either MEK1, ERK or SEK1, reduced small t-dependent induction of the cyclin D1 promoter. SV40 small t induction of the cyclin D1 promoter involves both the ERK and SAPK pathways that together may contribute to the proliferative and transformation enhancing activity of small t antigen.
The orchestration of orderly G1 phase progression requires temporally coordinated interactions between the cyclin-dependent kinases and their substrates, including the tumor suppressor protein pRB (1–3). The abundance of cyclin D1 appears to be rate-limiting in G1 phase progression in several different cell types containing pRB as evidenced by immunoneutralization and antisense experiments (4–9). The capacity of cyclin D1 to promote G1 phase progression likely relates to its function as the regulatory subunit of a holoenzyme that phosphorylates pRB and the related protein, p107 (1, 10). Cyclin D1 mRNA abundance is increased by the addition of growth factors and by overexpression of p21ras or p60src (11, 12). c-Jun induces the cyclin D1 promoter through an enhancer sequence at −953 (13) that binds c-Fos and c-Jun proteins (13, 14). In contrast, cyclin D1 mRNA levels were reduced in cell lines overexpressing c-Fos (15), and cotransfection of c-Fos expression vectors decreased cyclin D1 promoter activity in transient assays (13).
The early region of simian virus 40 (SV40) encodes large tumor (T) and the 20-kDa small tumor polypeptide (small t). SV40 small t antigen is required for the efficient transformation of several cell types especially when assayed in growth-arrested cells (16). SV40 infection is capable of stimulating quiescent cells to re-enter the S phase of the cell cycle (17–19). In addition, small t promotes G1 phase progression and S phase entry (20). The mechanisms by which small t enhances the transformation of resting cells may relate to its ability to stimulate or repress transcription of certain target genes (21–23). In addition, through inhibiting phosphatases that may down-regulate a proliferative signal, small t antigen may amplify and sustain normally transient signaling events (24–27).
One reported consequence of small t inhibition of PP2A is activation of members of the mitogen-activated protein kinase (MAPK) family (24). These are serine/threonine kinases that are induced by a wide variety of signaling pathways and may convey proliferative signals in G1 (28). Members of the MAPK family include the p42ERK and p44ERK, the p38 kinase (29), and a family of p54 kinases, the stress-activated protein kinases (SAPKs or Jun kinases) (30, 31). Activation of MAPKs requires dual phosphorylation on Thr and Tyr residues by MAPK kinases known as MEKs [ERK kinase (MAPKK), or SEK (SAPK kinase)] (28, 32, 33). Thus, small t antigen may maintain MAPK family members in more active states by preventing dephosphorylation of their Thr residues by PP2A. The activities of p42ERK and p44ERK are also down-regulated by dephosphorylation with the dual specificity phosphatase MKP-1 (34).
MAPKs augment the activity of several transcription factors including the activator protein-1 complex (AP-1, c-Fos/c-Jun), ATF-2, and members of the ETS transcription factor family. In some cell types, SV40 small t cooperates with ERK to stimulate AP-1 activity (24–26). PP2A was also identified as the primary phosphatase dephosphorylating the transcription factor CREB (cAMP-responsive element binding protein) in HepG2 cells (35), and overexpression of small t augmented CREB’s transcriptional activity in HepG2 cells (27). Although the enhancement of MAPK activity is associated with proliferative processes, target genes induced by small t that are capable of promoting G1 phase progression have yet to be identified.
In this report, we examined the role of small t antigen in regulating transcriptional activity of the cyclin D1 promoter. Small t activated the cyclin D1 promoter primarily through the c-Jun (AP-1) binding site at −953 with an additional contribution from a CREB binding site at −57. Mutation of domains involved in PP2A binding reduced small t activation of the cyclin D1 promoter. Induction of the cyclin D1 promoter by small t antigen through these two enhancer sequences may contribute to the proliferation- and transformation-enhancing functions of small t antigen.
MATERIALS AND METHODS
Construction of Plasmid Vectors.
A series of 5′ promoter reporter constructions derived from the human cyclin D1 genomic clone were recently described (13). The −163CD1LUC and −66CD1LUC reporters were constructed using 5′ amplimers directed to the published sequence. The Sp-1 site between −127 and −99 was deleted by PCR-directed mutagenesis in the context of the −163 bp promoter fragment to create −163ΔSp-1.
Cyclin D1 promoter enhancer sequences were linked to the thymidine kinase (TK81) promoter (TK81pA3LUC) to make CD1(AP-1)TKLUC (the cyclin D1 AP-1 site from −963 to −940), CD1(E2F)TKLUC (the cyclin D1 sequences from −156 to −133), and CD1(CRE)TKLUC (sequences from −66 to −40). The reporter p3TPLUX contains trimeric wild-type AP-1 responsive reporter elements (36), RSVLUC and pCMVLUC contain the viral promoters linked to the luciferase reporter, and the reporter (UAS)5E1BTATALUC (37) consists of multimerized GAL4 DNA binding sites cloned into the reporter pA3LUC.
Expression Vectors.
The SV40 small t expression plasmids pCMVt and pw2t, and the mutant small t plasmids pw2tP43L/K45N, pw2tC97S, pw2tC103S, pw2tP101A, pw2tC97S, and C103S were as described (38). The pw2t dl888, which lacks the small t splice donor, served as a control plasmid. The plasmids GAL4-c-Jun 5–253 (39) and murine (40) GAL4-CREB, the expression vectors pCMV-p41ERK (MAPKwt) and pCMV-p41ERKAla54 Ala55) (MAPKmut) (41), the dominant negative ERK expression vector (MAPKi) (42), the dominant negative MEK1 (Ala-218/Ala222) (referred to as MEKC) (43), the constitutively active MEK expression plasmids pCMV-MKKΔN3 and pCMV-MKKΔN3/S218E/S222D (44), the MKP-1 expression vector (34), and the dominant negative SEK mutants pEBG-SEK1-KR (SEKKR) and pcDNA3-SEK1-AL (SEKAL) were recently described (37, 45, 46).
Cell Culture and DNA Transfection.
Cell culture, DNA transfection, and luciferase assays were performed as previously described (36). The human trophoblast cell line JEG-3 and the fibroblast cell line CV-1 were maintained in DMEM with 10% fetal calf serum with 1% penicillin/streptomycin. Cells were transfected by calcium phosphate precipitation, the medium was changed after 6 hr, and the cells were processed for either luciferase activity or kinase activity at 24–48 hr. The fold effect was determined for a given construct by comparison with the effect of equal molar amounts of the expression vector cassette.
MEK, ERK, SAPK, and JNK1 Immune-Complex Kinase Assays.
JEG-3 cells were transfected with small t expression vectors for 48 hr, and then extracted with 20 mM Tris base (pH 7.8) containing 100 mM sodium chloride and 0.5% Nonidet P-40. Extracts were then incubated for 2 hr at 4°C with protein A-Sepharose beads that had been coated with antibodies to MEK1 (Transduction Laboratories, Lexington, KY), ERK (C16) and JNK1 (C17) (Santa Cruz Biotechnology), and SAPK (30). Immunoprecipitates were then washed once with extraction buffer, twice with 0.5 M lithium chloride in 0.1 M Tris base (pH 8.6), and then once with the kinase buffer (25 mM Hepes, pH 7.4/10 mM magnesium chloride/10 mM manganese chloride/1 mM dithiothreitol). Kinase reactions were performed at room temperature for 20 min in 30 μl buffer containing 10 μCi [γ-32P]ATP (3000 Ci/mmol; 1 Ci = 37 GBq) and 1–2 μg of appropriate substrate proteins. These were myelin basic protein for ERK kinase, histidine-tagged kinase inactive ERK for MEK1 (47), and GST c-Jun (1) for JNK1 (48) and SAPK (30). Aliquots of each reaction were quantitated by acid precipitation. MEK1 reactions were spotted on P81 cellulose (Whatman), and then washed with phosphoric acid before scintillation counting. For ERK and JNK1 assays, aliquots of the reactions were diluted to 0.5 ml with water containing 100 μg carrier bovine serum albumin, and then precipitated at 4°C using 10% (JNK1) or 30% (ERK) trichloroacetic acid. In most experiments, results obtained by acid precipitation were confirmed by analysis of a second aliquot of each reaction on SDS/polyacrylamide gels, followed by autoradiography to visualize reaction products.
Electrophoretic Mobility-Shift Assays.
Electrophoretic mobility-shift assays were performed using nuclear extracts (5–10 μg) from JEG-3 cells and binding buffer containing 20 mM Tris·Cl (pH 7.8), 100 mM KCl, 1.0 mM EDTA, and 10% glycerol to which 0.5 ng of γ-32P-labeled probe and 50 μg/ml poly(dI·dC) as competitor (36, 49). The sequence of the cyclin D1 promoter CRE site oligodeoxyribonucleotides (CD1CREwt) was 5′-AAC AAC AGT AAC GTC ACA CGG AC-3′ and the CD1CREmut sequence was 5′-AAC AAC AGT cgC GTC ACc CGG AC-3′. The protein–DNA complexes were analyzed by electrophoresis through a 5% polyacrylamide gel, with 0.5 × Tris·borate, EDTA buffer (TBE buffer: 0.045 M Tris·borate/0.001 M EDTA). Supershifts were performed with antibodies to c-Jun (KM-1X), ATF-2 (C-19X), c-Fos (K-25X), (Santa Cruz Biotechnology), and to CREB/CREM (HM93) (50). The gels were dried and exposed to XAR5 radiographic film.
Western Blots.
The abundance of cyclin D1 protein was determined by Western blotting using the monoclonal cyclin D1 antibody (HD-11; Santa Cruz Biotechnology) and an anti-mouse horseradish peroxidase second antibody (14). The Enhanced Chemiluminescence system (Amersham) was used to visualize reactive proteins, and densitometry of the autoradiograms was performed using the Bio-Rad molecular analyst software (version 1.01).
RESULTS
SV40 Small t Activates Cyclin D1 Promoter Activity.
The effect of SV40 small t on cyclin D1 promoter activity was examined by cotransfection experiments using the −1745CD1LUC reporter and the SV40 small t expression plasmids (pw2t and CMVt). Induction of the −1745CD1LUC reporter increased with the amount of transfected expression plasmid (Fig. 1A). Cyclin D1 reporter activity was induced 6- to 7-fold in JEG-3 cells (Fig. 1A). Several native and synthetic promoters were examined to further elucidate the specificity of the SV40 small t-dependent induction of the cyclin D1 promoter. The minimal TK81 promoter and pA3LUC were not affected by SV40 small t expression (not shown).
Figure 1.
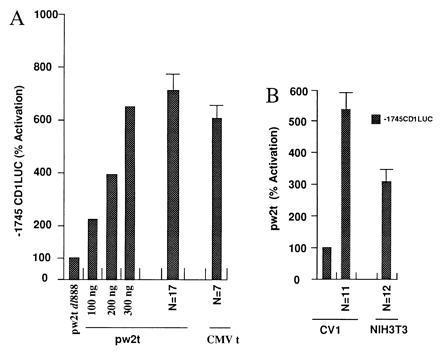
The cyclin D1 promoter is activated by SV40 small t. (A) The −1745CD1LUC reporter was transfected with either the control plasmid pw2t dl888 or the small t expression vector. For the dose response, a representative example from three separate experiments is shown for increasing amounts of transfected expression plasmid normalized to the effect of vector alone. For the effect of pw2t (n = 17 separate transfections) or the CMV small t expression plasmid (n = 7), the data are expressed as mean ± SEM. (B) The −1745CD1LUC reporter was transfected with the SV40 small t expression vector in the cell lines indicated. The data are shown expressed as mean ± SEM with the number of experiments shown as N.
To determine whether SV40 small t was capable of regulating the cyclin D1 promoter in different cell types, transfections were conducted in CV-1 and NIH 3T3 cells. Overexpression of small t activated the cyclin D1 promoter 5-fold in CV-1 cells and 3- to 4-fold in NIH 3T3 cells (Fig. 1B).
The Cyclin D1 AP-1 Site and CRE Site Are Required for Full Activation by Small t.
The regions of the cyclin D1 promoter involved in transcriptional induction by small t were explored using a series of cyclin D1 5′ promoter constructions transfected in conjunction with either the wild-type SV40 small t vector or the dl888 control (Fig. 2A). The induction of cyclin D1 transcription by pw2t was reduced 80% upon deletion from −964 to −161 (Fig. 2A). A sequence at −953 resembles the collagenase AP-1 site and was previously shown to convey induction of the cyclin D1 promoter by c-Jun (13). Site-directed mutation of the AP-1 site, previously shown to abolish binding of AP-1 proteins in electrophoretic mobility-shift assays (13), in the context of the −964-bp promoter fragment (−964 mtCD1LUC), reduced small t transactivation by 75%. The proximal promoter fragments −141CD1LUC and −66CD1LUC, maintained a 2- to 3-fold induction by small t (Fig. 2A), whereas the −22CD1LUC reporter and the plasmid vector pA3LUC were not induced significantly by small t compared with the dl888 control. These findings indicated that the additional small t-responsive sequences of the cyclin D1 promoter were located between −66 and −22, a region that contains a CRE/ATF recognition sequence.
Figure 2.
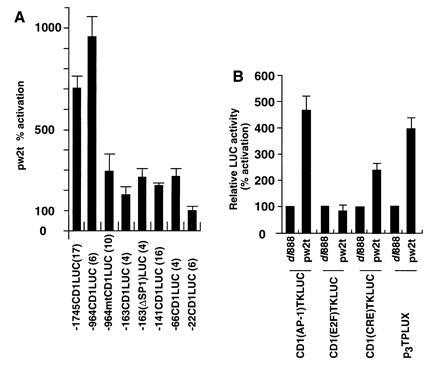
The −953 region of the cyclin D1 promoter is required for activation by small t. (A) A series of 5′ promoter deletions of the cyclin D1 promoter were transfected into JEG-3 cells with the small t expression plasmid. The data are the mean fold induction ± SEM by pw2t determined by comparison with cells transfected with the “cassette” dl888. Data is for N separate transfections as indicated in parenthesis. (B) The cyclin D1(AP-1)TKLUC reporter was transfected with small t into JEG-3 cells. Data are normalized for the effect of the pw2t vector (shown as 1). Comparison was made with the effect of small t on the synthetic reporters, CD1(E2F)TKLUC, CD1(CRE)TKLUC (encoding the sequences resembling a putative CRE binding site at −57), and the synthetic AP-1 site reporter p3TPLUX. The data are shown expressed as mean ± SEM.
To evaluate the ability of small t to affect the cyclin D1 AP-1 and CRE/ATF site, transient expression studies were performed with synthetic constructions in which these sites were linked to the minimal TK promoter. The CD1(AP-1)TKLUC reporter was induced 4- to 5-fold by small t (Fig. 2B). The element resembling the E2F site [CD1(E2F)TKLUC], was not induced by small t. The cyclin D1 CRE site at −57 was sufficient for a 3-fold induction by small t (Fig. 2B). The synthetic multimeric collagenase AP-1 site reporter, p3TPLUX, was also induced 4-fold by small t (Fig. 2B). Together these studies demonstrate that primarily the AP-1 site but also the CRE/ATF site of the cyclin D1 promoter convey induction by small t antigen.
The PP2A Binding Domain of Small t Is Required for Full Activation of Cyclin D1 Expression.
To determine the domains of small t required for activation of the cyclin D1 promoter, a series of plasmids expressing either wild-type or mutant small t proteins was examined in conjunction with the −1745CDLUC reporter in JEG-3 cells (Fig. 3). Previous studies demonstrated that site-directed mutagenesis of small t amino acids 97 (pw2tC97S) and 103 (pw2tC103S) or 101 (pw2tP101A) impaired the binding of small t to PP2A (38) without affecting the stability of small t protein. Transactivation of the adenovirus E2 promoter was not affected by mutations in the 97–103 region, but was eliminated by a double mutation in another region of small t, P43L/K45N. Mutation of amino acids in the 97–103 PP2A binding region reduced small t transactivation of the cyclin D1 promoter by 50–60% in JEG-3 cells (Fig. 3A). Mutations at amino acids 43 and 45 (pw2tP43L/K45N) resulted in a slight enhancement of cyclin D1 promoter activity compared with the wild type (Fig. 3A).
Figure 3.
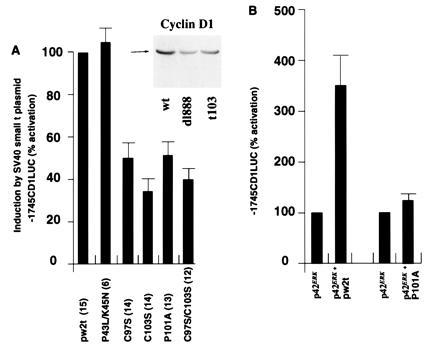
The domains of small t required for activation of the cyclin D1 promoter. (A) The −1745CD1LUC reporter was cotransfected with expression vectors encoding either wild-type or mutant SV40 small t proteins into JEG-3 cells. The data shown were adjusted for the effect of expression vector cassette lacking the cDNA on −1745CD1LUC reporter activity. The mean data (± SEM) of n separate transfections (in parenthesis) are shown. (Inset) Western blot of JEG-3 cells transfected with the wild-type small t plasmid, the dl888, or the PP2A binding defective mutant t103 probed with the cyclin D1 antibody. (B) As overexpression of ERK was previously shown to augment activity of the cyclin D1 promoter in JEG-3 cells (13), the −1745CD1LUC reporter was cotransfected with the expression vector encoding wild-type p42ERK in conjunction with either the wild-type or mutant small t proteins into JEG-3 cells. The data were normalized to 1 for the effect of p42ERK on the −1745CD1LUC reporter. The mean data (± SEM) of 7 to 8 separate transfections are shown.
Findings with transient transcription assays were reflected in levels of endogenous cyclin D1 in transfected cells. Cells transfected with pw2t had elevated levels of cyclin D1 (Fig. 3A Inset). These levels under represent effects of small t antigen. Although transfection efficiencies reach up to 40% in JEG-3 cells, there is a significant number of untransfected cells in these experiments. Less cyclin D1 was present in cells transfected by pw2tC103S, suggesting that the increase in cyclin D1 protein levels required, at least in part, the inhibition of PP2A. In separate transfections, the mean induction of cyclin D1 protein in JEG-3 cells transfected with wild-type small t was 1.9-fold greater than in cells transfected with the pw2tC103S mutant (n = 5) (data not shown).
It had been shown previously that overexpression of ERK induced the cyclin D1 promoter 3- to 4-fold in JEG-3 cells (13). As an additional assay of PP2A inhibition by small t, the wild-type and pw2tP101A expression vectors were transfected with the cyclin D1 promoter in the presence of the expression vector for p42ERK (Fig. 3B). The wild type but not the mutant small t antigen augmented transactivation of the cyclin D1 promoter by p42ERK (Fig. 3B), consistent with the prediction that small t would increase ERK activity through inhibition of PP2A.
Kinase Activities in Small t Transfected Cells.
Previous studies in monkey kidney CV-1 cells suggested that small t antigen could activate ERK and MEK, most likely by inhibition of PP2A (24). Similarly, levels of ERK and MEK were examined in JEG-3 cells that were transfected with wild-type or mutant small t expression plasmids for 48 hr. As shown in Fig. 4A, ERK activity was induced 5-fold by small t antigen, an effect that was eliminated by a mutation in the PP2A binding region, pw2tC103S, but not by mutation of residues 43 and 45. Similarly, MEK1 activity was increased 3-fold by small t antigen in a PP2A-dependent fashion (Fig. 4B). Because activation of the cyclin D1 promoter by small t occurred mainly through an AP-1 site, JNK1 activity was also studied. Small t stimulated JNK1 activity (Fig. 4C) and SAPK activity 2.5-fold (not shown), and this stimulation was decreased by the pw2tC103S mutation.
Figure 4.
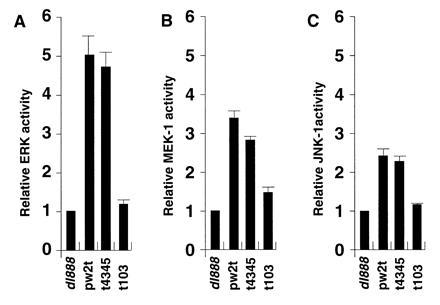
SV40 small t stimulates ERK, MEK, and JNK activity in JEG-3 cells. JEG-3 cells were transfected with a plasmid that expressed wild-type small t antigen (pw2t), a small t mutant with reduced PP2A binding (t103), a small t mutant that does not affect PP2A binding (t4345), or no small t antigen (dl888). Cells were extracted 48 hr after transfection and kinase assays were done on immunoprecipitates obtained with antibodies against ERK (A), MEK1 (B), or JNK1 (C). Incorporation of [γ-32P] into substrates myelin basic protein (A), kinase-inactive ERK (B), and GST c-Jun (C) was quantitated by acid precipitation and liquid scintillation counting as described. The data are expressed as a ratio between small t-induced kinase activity and that of the dl888 negative control.
Small t Induction of the Cyclin D1 Promoter Requires MEK, ERK, and SAPK Pathways.
As MEK activity was induced in small t-transfected JEG-3 cells, experiments were conducted to determine whether MEK was required for full induction of the cyclin D1 promoter by the small t antigen. The catalytically active MEK mutants MEKΔN3 and MEKΔN3/S218E/S222D (44) induced both the p3TPLUX and the −1745 CD1LUC reporters 4- to 5-fold (Fig. 5A). Small t-dependent induction of the cyclin D1 promoter was reduced 30% by overexpression of the dominant negative MEK expression vector, MEKC (Fig. 5B), 40% by the dominant negative ERK expression plasmid (MAPKi), and 60% by the addition of the MEK and ERK inhibitor PD098059 (20 μM) (51) (Fig. 5B). Overexpression of the dominant negative SEK expression vectors, SEK1KR, and SEK1AL (45, 46), reduced small t induced promoter activity by 30%, indicating that both ERK and SEK pathways contribute to the induction of the cyclin D1 promoter by small t antigen. Overexpression of the dual specificity phosphatase MKP-1 reduced small t induced cyclin D1 promoter activity 90% (Fig. 5B).
Figure 5.
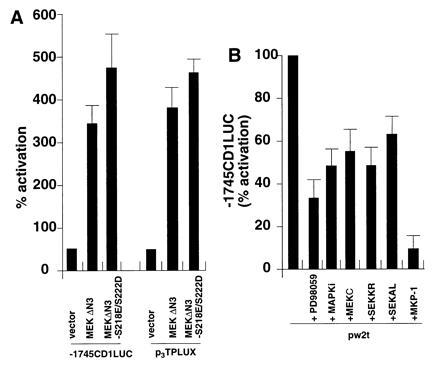
MEK, ERK, and SAPK are required for full induction of the cyclin D1 promoter by small t antigen. JEG-3 cells were transfected with either (A) the activating MEK mutants, MEKΔN3, and MEKΔN3 S218E/S222D or (B) wild type small t and the expression plasmids encoding either dominant negative mutants of MEK (MEKC), p42ERK (MAPKi), SEK, or the MKP-1 expression vector encoding a dual specificity Thr/Tyr phosphatase.
c-Jun and CREB Transactivation Functions Are Induced by Small t.
The transient expression studies indicated that both the AP-1 site and a CRE/ATF site contributed to the full induction of the cyclin D1 promoter by small t antigen. Direct effects of small t on c-Jun activity were studied because it was known that c-Jun was one of the proteins that bound the cyclin D1 AP-1 site (13) and that small t could increase JNK1 activity in transfected cells. As shown in Fig. 6A, small t induced c-Jun activity, as studied using a construct that expressed a chimeric GAL4-c-Jun protein and a reporter gene that contained GAL4 binding sites (Fig. 6A). Small t had no effect on the (UAS)5E1BTATALUC reporter.
Figure 6.
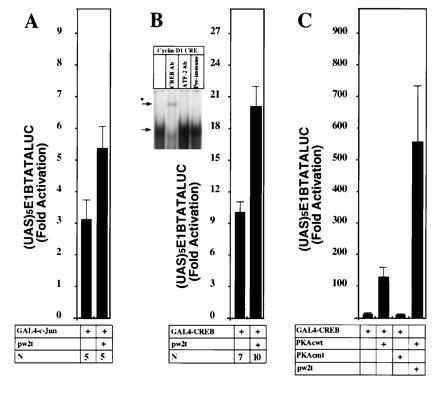
Small t augments c-Jun and CREB transactivation in JEG-3 cells. (A) The GAL4-c-Jun construct (1.2 μg) was transfected with the small t expression plasmids (600 ng) and the (UAS)5E1BTATALUC reporter (4.8 μg) in JEG-3 cells. The mean data (± SEM) of n separate transfections are shown. (Comparison is shown normalized for the effect of dl888). (B Inset) An electrophoretic mobility-shift assay was performed with the cyclin D1 CRE/ATF site using nuclear extracts from JEG-3 cells, with the addition of specific antibodies as described. The arrow indicates the primary complex formed and the asterisk indicates the supershifted complex formed with the CREB/CREM antibody HM93. (B) The GAL4-CREB construct (1.2 μg) was transfected with the small t expression plasmid (600 ng) and (UAS)5E1BTATALUC (4.8 μg) either alone or with the addition of an expression plasmid for wild-type or mutant protein kinase A catalytic subunit (C).
The cyclin D1 CRE site was found to bind a JEG-3 cell nuclear complex that was supershifted with antibody to CREB/CREM protein but not with antibody to c-Fos, c-Jun, or ATF-2 (Fig. 6B Inset). Accordingly, the effect of small t on CREB transactivation function was examined, using a GAL4-CREB fusion protein and a GAL4 responsive reporter plasmid. Like a wild-type PKA catalytic subunit, small t induced activity of CREB-GAL4 protein (Fig. 6 B and C). It is interesting to note that small t augmented the PKA catalytic subunit activation of CREB-GAL4.
DISCUSSION
The ability of SV40 infection to stimulate quiescent cells to reenter the cell cycle has been known for some time (17–19). The SV40 small t antigen is also known to stimulate cell-cycle progression (17, 19, 20). In nonpermissive infection of murine cells (18) and in permissive infection of monkey cells (20), small t antigen promotes G1 phase progression. In semipermissive human cells, small t is required for the induction of focus formation (52). As recent studies demonstrated that small t antigen may induce cyclin A expression (53), small t antigen appears to induce several components of the G1 and S phase cell-cycle regulatory apparatus. Our studies strongly implicate the inhibition of PP2A and both an AP-1 site at −953 and a CRE site at −57 for full activation of cyclin D1 transcription by SV40 small t.
The ability of small t antigen to transactivate promoters in transient transfections has been described previously, primarily using the adenovirus E2A promoter (AdE2A) as a model (22). As with cyclin D1, an ATF/CRE site in the AdE2A promoter was involved in small t responsiveness. Deletion of the ATF/CRE site did not eliminate small t transactivation; however, the overall response to small t was significantly reduced in magnitude. In contrast, deletion of the two E2F sites in the E2A promoter eliminated transactivation by small t antigen (21), whereas the E2F site in the cyclin D1 promoter was not necessary in this study. In addition, the transactivation of E2A did not require the inhibition of PP2A by small t antigen and was eliminated in small t that carried double mutations at residues 43 and 45 (38). The same mutations had no effect on transactivation of the cyclin D1 promoter.
Point mutations in the 97–103 PP2A binding region of small t reduced the ability of small t to transactivate the cyclin D1 promoter in JEG-3 cells. Consistent with earlier studies in CV-1 cells, inhibition of PP2A by small t also correlated with reduced ability of the mutant proteins to increase activities of MAPK family members in transfected JEG-3 cells. Direct evidence for a role for MAPK family members in transcriptional activation of cyclin D1 was obtained by transfection of p42ERK. Small t augmented the transcriptional activation by p42ERK, an effect that was also dependent on PP2A inhibition. In addition, constructs that expressed dominant-negative forms of several of the MAPKs reduced small t activation of the cyclin D1 promoter.
A new finding of the current study is the activation of JNK1 by small t antigen. As with the induction of MEK and ERK, the induction of JNK required the PP2A binding domains of small t. Dominant-negative SEK constructs reduced activation of cyclin D1 promoter activity by small t. The combined activities of SAPK (JNK) and of MEK/ERK would be likely to increase cellular AP-1 activity through phosphorylation of c-Jun (31, 54), a factor known to bind the cyclin D1 AP-1 site (13). In fact, mutation of the AP-1 site at −953 dramatically reduced transactivation by small t, and, in these studies, small t induced both the collagenase and cyclin D1 AP-1 sites in heterologous constructs. In our studies, c-Jun transactivation function, as assessed by GAL4 heterologous reporter assays, was induced by small t, suggesting that a component of small t-dependent induction of the cyclin D1 promoter may be mediated by this mechanism. Activation of AP-1 by small t supports a previous report that small t could induce a synthetic AP-1 reporter in microinjected REF52 and CV-1 cells in a PP2A-dependent fashion (25). In contrast, small t inhibited AP-1 activity in transfected CV-1P cells (23), raising the possibility that there are cell-type specific differences in intracellular signaling by small t antigen.
Induction of cyclin D1 promoter activity was not totally eliminated by mutation of the AP-1 site and residual activity correlated with the presence of the CRE site at −57. Additional evidence for the involvement of the CRE site include the induction by small t of the cyclin D1 CRE site when linked to the minimal TK promoter and the induction of CREB transactivation function by small t. Although not formally shown, it is likely that the effect of small t on CREB, as on AP-1, is via the inhibition of PP2A.
Although increased MEK and ERK activity appear to contribute significantly to the induction of cyclin D1 promoter activity, small t mutants defective in PP2A binding maintained residual activation of the promoter. Interestingly, sequences in the amino-terminal region of small t are involved in transactivation of the AdE2A and cyclin A genes (38, 53), as well as the repression of c-fos transcription (23). The amino-terminal region is shared by small t and the viral transforming protein large T, raising the question of individual effects of the two viral proteins on transcription of cellular target genes. It has been suggested that the expression of SV40 large T, like the adenovirus E1A protein, may reduce cyclin D1 levels by binding the Rb protein, a positive regulator of cyclin D1 transcription (55). Such down-regulation of cyclin D1 by T antigen may occur at late times in the course of an infection, a period when cyclin D1 expression would no longer be necessary or advantageous. At early times, cyclin D1 expression would stimulate progression of infected cells through G1, a process known to be enhanced by small t (18, 20). It is also interesting to note that the shared domain of T/t has functional similarity to the amino terminal domain of E1A which has transcriptional regulatory activities as well. In genetic complementation studies, this region of T/t could provide function to amino terminal mutants of E1A (56, 57).
It was also intriguing that the activation of the cyclin D1 promoter by small t was reduced by MKP-1. MKP-1 is encoded by the mitogen-inducible gene 3CH134 and inactivates p42ERK and p44ERK by dephosphorylating both phosphothreonine and phosphotyrosine regulatory sites (58). Microinjection of MKP-1 blocked S phase entry induced by ERKs (34). Our studies indicate that MKP-1 may have an important effect in regulating transcriptional activity of the cyclin D1 gene. It will be of interest to determine whether a reduction in cyclin D1 expression by MKP-1 contributes to the cell-cycle inhibitory effect of this phosphatase (34).
Acknowledgments
We are grateful to Drs. J. Massague, B. Thimmapaya, R. Tjian, N. Ahn, L. Lau, F. McCormick, R. Goodman, G. Johnson, S. McDonald, and R. Davis for plasmids and Dr. J. Habener for the CREB/CREM antibody HM93. The reagent PD098059 was supplied by Dr. A. Saltiel from Parke Davis. This work was supported in part by the National Cancer Institute Grants R29CA70896-01 (R.G.P.) and CA21327 (K.R.); the Aichi Health Promotion Foundation, Owari Kenyu-kai Committee and the Takasu Foundation (G.W.); National Institutes of Health Training Grants 5 T32 GM0852-10 (R.J.L.) and CA09560 (A.N.K.); and a David Shemin Undergraduate Fellowship from Northwestern University (I-W.S.).
Footnotes
Abbreviations: SV40, simian virus 40; small t antigen, small tumor antigen; MAPK, mitogen-activated protein kinase; ERK, extracellular signal regulated kinase; SAPK, stress-activated protein kinase; SEK, SAPK kinase; CREB, cAMP-responsive element binding protein.
References
- 1.Weinberg R A. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 2.Sherr C J, Roberts J M. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 3.Sherr C J. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 4.Baldin V, Lukas J, Marcote M J, Pagano M, Draetta G. Genes Dev. 1993;7:812–821. doi: 10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- 5.Quelle D E, Ashmun R A, Shurtleff S A, Kato J-y, Bar-Sagi D, Roussel M F, Sherr C J. Genes Dev. 1993;7:1559–1571. doi: 10.1101/gad.7.8.1559. [DOI] [PubMed] [Google Scholar]
- 6.Jiang W, Kahn S M, Zhou P, Zhang Y-J, Cacace A M, Infante A S, Doi S, Santella R M, Weinstein I B. Oncogene. 1993;8:3447–3457. [PubMed] [Google Scholar]
- 7.Resnitzky D, Gossen M, Bujard H, Reed S I. Mol Cell Biol. 1994;14:1669–1679. doi: 10.1128/mcb.14.3.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Musgrove E A, Lee C S, Buckley M F, Sutherland R L. Proc Natl Acad Sci USA. 1994;91:8022–8026. doi: 10.1073/pnas.91.17.8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartkova J, Lukas J, Muller H, Lutzhoft D, Strauss M, Bartek J. Int J Cancer. 1994;57:351–361. doi: 10.1002/ijc.2910570311. [DOI] [PubMed] [Google Scholar]
- 10.Beijersbergen R L, Carlee L, Kerkhoven R M, Bernards R. Genes Dev. 1995;9:1340–1353. doi: 10.1101/gad.9.11.1340. [DOI] [PubMed] [Google Scholar]
- 11.Bodrug S, Warner B J, Bath M L, Lindeman G J, Harris A W, Adams J M. EMBO J. 1994;13:2124–2130. doi: 10.1002/j.1460-2075.1994.tb06488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J-J, Chao J-R, Jiang M-C, Ng S-Y, Yen J J-Y, Yang-Yen H-F. Mol Cell Biol. 1995;15:3654–3663. doi: 10.1128/mcb.15.7.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell R G. J Biol Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe G, Lee R J, Albanese C, Rainey W E, Batlle D, Pestell R G. J Biol Chem. 1996;271:22570–22577. doi: 10.1074/jbc.271.37.22570. [DOI] [PubMed] [Google Scholar]
- 15.Miao G M, Curran T. Mol Cell Biol. 1994;14:4295–4310. doi: 10.1128/mcb.14.6.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin R G, Setlow V P, Edwards C A F, Vembu D. Cell. 1979;17:635–643. doi: 10.1016/0092-8674(79)90271-x. [DOI] [PubMed] [Google Scholar]
- 17.Henry P P, Black H, Oxman M N, Weissman S M. Proc Natl Acad Sci USA. 1966;56:1170–1176. doi: 10.1073/pnas.56.4.1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiscott J B, Defendi V. J Virol. 1981;37:802–812. doi: 10.1128/jvi.37.2.802-812.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith H S, Scher C D, Todaro G J. Virology. 1971;44:359–370. doi: 10.1016/0042-6822(71)90267-4. [DOI] [PubMed] [Google Scholar]
- 20.Cicala C, Avanaggiati M L, Graessmann A, Rundell K, Levine A S, Carbone M. J Virol. 1994;68:3138–3144. doi: 10.1128/jvi.68.5.3138-3144.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loeken M R. J Virol. 1992;66:2551–2555. doi: 10.1128/jvi.66.4.2551-2555.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loeken M, Bikel I, Livingstone D M, Brady J. Cell. 1988;55:1171–1177. doi: 10.1016/0092-8674(88)90261-9. [DOI] [PubMed] [Google Scholar]
- 23.Wang W-B, Bikel I, Marsilio E, Newsome D, Livingston D M. J Virol. 1994;68:6180–6187. doi: 10.1128/jvi.68.10.6180-6187.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sontag E, Fedorov S, Kamibayashi C, Robbins D, Cobb M, Mumby M. Cell. 1993;75:887–897. doi: 10.1016/0092-8674(93)90533-v. [DOI] [PubMed] [Google Scholar]
- 25.Frost J A, Alberts A S, Sontag E, Guan K, Mumby M C, Feramisco J R. Mol Cell Biol. 1994;14:6244–6252. doi: 10.1128/mcb.14.9.6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alberts A S, Deng T, Lin A, Meinkoth J L, Schonthal A, Mumby M C, Karin M, Feramisco J R. Mol Cell Biol. 1993;13:2104–2112. doi: 10.1128/mcb.13.4.2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wheat W H, Roesler W J, Klemm D J. Mol Cell Biol. 1994;14:5881–5890. doi: 10.1128/mcb.14.9.5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cobb M H, Goldsmith E J. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 29.Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, Hunt T, Nebreda A R. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 30.Kyriakis J M, Banerjee P, Nikolakaki E, Dai T, Rubie E A, Ahmad M F, Avruch J, Woodgett J R. Nature (London) 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 31.Minden A, Lin A, Smeal T, Derijard B, Cobb M, Davis R, Karin M. Mol Cell Biol. 1994;14:6683–6688. doi: 10.1128/mcb.14.10.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng C F, Guan K L. J Biol Chem. 1993;268:11435–11439. [PubMed] [Google Scholar]
- 33.Wu J, Harrison J K, Vincent L A, Haystead C, Haystead T A, Michel H, Hunt D F, Lynch K R, Sturgill T W. Proc Natl Acad Sci USA. 1993;90:173–177. doi: 10.1073/pnas.90.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun H, Tonks N K, Bar-Sagi D. Science. 1994;266:285–288. doi: 10.1126/science.7939666. [DOI] [PubMed] [Google Scholar]
- 35.Wadzinski B E, Wheat W H, Jaspers S, Peruski L F J, Lickteig R L, Johnson G L, Klemm D J. Mol Cell Biol. 1993;13:2822–2834. doi: 10.1128/mcb.13.5.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pestell R G, Albanese C, Hollenberg A, Jameson J L. J Biol Chem. 1994;269:31090–31096. [PubMed] [Google Scholar]
- 37.Pestell R G, Albanese C, Watanabe G, Lee R J, Lastowiecki P, Zon L I, Ostrowski M, Jameson J L. Mol Endocrinol. 1996;10:1084–1094. doi: 10.1210/mend.10.9.8885243. [DOI] [PubMed] [Google Scholar]
- 38.Mungre S, Enderle K, Turk B, Porras A, Wu Y Q, Mumby M C, Rundell K. J Virol. 1994;68:1675–1681. doi: 10.1128/jvi.68.3.1675-1681.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baichwal V R, Park A, Tjian R. Nature (London) 1991;352:165–168. doi: 10.1038/352165a0. [DOI] [PubMed] [Google Scholar]
- 40.Gonzalez G A, Montminy M R. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 41.Seth A, Gonzalez F A, Guta S, Raden D L, Davis R J. J Biol Chem. 1992;267:24796–24804. [PubMed] [Google Scholar]
- 42.Conrad K E, Oberwetter J M, Vaillancourt R, Johnson G L, Gutierrez-Hartmann A. Mol Cell Biol. 1994;14:1553–1565. doi: 10.1128/mcb.14.3.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiu R G, Chen J, Kirn D, McCormick F, Symons M. Nature (London) 1995;374:457–459. doi: 10.1038/374457a0. [DOI] [PubMed] [Google Scholar]
- 44.Mansour S J, Matten W T, Hermann A S, Candia J M, Rong S, Fukasawa K, Vande Woude G F, Ahn N G. Science. 1994;265:966–969. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 45.Yan M, Dai T, Deak J C, Kyriakis J M, Zon L I, Woodgett J R, Templeton D J. Nature (London) 1994;372:798–800. doi: 10.1038/372798a0. [DOI] [PubMed] [Google Scholar]
- 46.Pombo C M, Kehrl J H, Sanchez I, Katz P, Avruch J, Zon L I, Woodgett J R, Force T, Kyriakis J M. Nature (London) 1995;377:750–754. doi: 10.1038/377750a0. [DOI] [PubMed] [Google Scholar]
- 47.Lange-Carter C A, Pleiman C M, Gardner A M, Blumer K J, Johnson G L. Science. 1993;260:315–319. doi: 10.1126/science.8385802. [DOI] [PubMed] [Google Scholar]
- 48.Derijard B, Hibi M, Wu I H, Barrett T, Su B, Deng T, Karin M, Davis R J. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 49.Pestell R G, Albanese C, Watanabe G, Johnson J, Eklund N, Lastowiecki P, Jameson J L. J Biol Chem. 1995;270:18301–18308. doi: 10.1074/jbc.270.31.18301. [DOI] [PubMed] [Google Scholar]
- 50.Waeber G, Meyer T E, LeSieur M, Hermann H L, Gerard N, Habener J F. Mol Endocrinol. 1991;5:1418–1430. doi: 10.1210/mend-5-10-1418. [DOI] [PubMed] [Google Scholar]
- 51.Pang L, Sawada T, Decker S J, Saltiel A R. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- 52.De Ronde A, Sol C J A, van Strien A, ter Schegget J, van der Noordaa J. Virology. 1989;171:260–263. doi: 10.1016/0042-6822(89)90534-5. [DOI] [PubMed] [Google Scholar]
- 53.Porras A, Bennett J, Howe A, Tokos K, Bouck N, Henglein B, Sathyamangalam S, Thimmapaya B, Rundell K. J Virol. 1996;70:6902–6908. doi: 10.1128/jvi.70.10.6902-6908.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karin M. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 55.Lukas J, Muller H, Bartkova J, Spitkovsky D, Kjerulff A A, Jansen-Durr, Srauss M, Bartek J. J Cell Biol. 1994;125:625–638. doi: 10.1083/jcb.125.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peden K W C, Pipas J M. Virus Genes. 1992;6:107–118. doi: 10.1007/BF01703060. [DOI] [PubMed] [Google Scholar]
- 57.Yaciuk P, Carter M C, Pipas J M, Moran E. Mol Cell Biol. 1991;11:2116–2124. doi: 10.1128/mcb.11.4.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun H, Charles C H, Lau L F, Tonks N K. Cell. 1993;75:487–493. doi: 10.1016/0092-8674(93)90383-2. [DOI] [PubMed] [Google Scholar]