

Abstract
Ro09-0198 is a tetracyclic polypeptide of 19 amino acids that recognizes strictly the structure of phosphatidylethanolamine (PE) and forms a tight equimolar complex with PE on biological membranes. Using the cyclic peptide coupled with fluorescence-labeled streptavidin, we have analyzed the cell surface localization of PE in dividing Chinese hamster ovary cells. We found that PE was exposed on the cell surface specifically at the cleavage furrow during the late telophase of cytokinesis. PE was exposed on the cell surface only during the late telophase and no alteration in the distribution of the plasma membrane-bound cyclic peptide was observed during the cytokinesis, suggesting that the surface exposure of PE reflects the enhanced scrambling of PE at the cleavage furrow. Furthermore, cell surface immobilization of PE induced by adding the cyclic peptide coupled with streptavidin to prometaphase cells effectively blocked the cytokinesis at late telophase. The peptide-streptavidin complex treatment had no effect on furrowing, rearrangement of microtubules, and nuclear reconstitution, but specifically inhibited both actin filament disassembly at the cleavage furrow and subsequent membrane fusion. These results suggest that the redistribution of the plasma membrane phospholipids is a crucial step for cytokinesis and the cell surface PE may play a pivotal role in mediating a coordinate movement between the contractile ring and plasma membrane to achieve successful cell division.
Keywords: phospholipid asymmetry, cell division, cytokinesis, contractile ring, actin filament
Mitotic phase of the cell cycle ends as the cytoplasmic components are divided by the process of cytokinesis. During cytokinesis, cell plasma membranes are drawn inward to form cleavage furrows, which gradually deepen and fuse with each other, resulting in cell division. Regarding the molecular mechanisms of cytokinesis, investigators have concentrated on the “sliding-filament contractile-ring” model (1–3). The contractile ring is a transient bipolar array of actin filaments with their barbed ends attached to the plasma membrane at sites around the equator of the dividing cell. The interaction of actin filaments with bipolar myosin II filaments applies tension to the membrane to form cleavage furrow, much like the contraction of smooth muscle cells. In this model, the reorganization of the cortical filaments plays a dominant role in the process, and no attention has been paid to the organization and function of membrane lipids.
The cytokinetic process ends with fusion of adjacent plasma membranes, which accompanies the dynamic rearrangement of architecture of membrane phospholipids (4). Phosphatidylethanolamine (PE), a dominant aminophospholipid of most living organisms, is a unique phospholipid in that it forms a nonbilayer structure, which facilitates rapid phospholipid trans-bilayer movement leading to membrane fusion in model membranes (5, 6). In various eukaryotic plasma membranes, aminophospholipids such as PE and phosphatidylserine reside in the inner leaflet, while choline-containing phospholipids such as phosphatidylcholine and sphingomyelin are localized mainly in the outer leaflet (7–9). It has been shown that PE concentration in the outer leaflet of the plasma membrane of myoblast increase prior to cell–cell fusion to form myotubes (10, 11). Erythrocytes that have lost the asymmetric distribution of phospholipids were shown to fuse more readily than those exhibiting the asymmetric distribution (12, 13). In dividing Chinese hamster ovary (CHO) cells, the amount of PE in the outer leaflet of plasma membrane increased by threefold when the cells entered the late telophase/G1 phase (14). Although these results suggest surface exposure of PE in late telophase or G1 phase, distribution of PE molecules on the cell surface and its role in the cytokinetic process remains unknown.
In this study, we have developed a novel peptide probe that binds specifically to PE in biological membranes. Ro09-0198 (Ro) is a 19-amino acid tetracyclic polypeptide with a molecular mass of 2041 Da isolated from Streptoverticillium griseoverticillatum. (15–17). We have shown that Ro strictly recognizes the structure of PE and forms a tight equimolar complex with PE on biological membranes (15–17). We have conjugated biotinylated Ro to fluorescence-labeled streptavidin (SA) to study the molecular movement of PE in dividing CHO cells. The analyses have shown that the fluorescence-labeled peptide bound specifically to cleavage furrow during late telophase of cytokinesis and the addition of the peptide complexed with SA to the mitotic cells effectively inhibited the cytokinesis at the late telophase. The peptide-SA complex treatment had no effect on furrowing, rearrangement of microtubules, and nuclear reconstitution, but specifically inhibited both actin filament disassembly at the cleavage furrow and subsequent membrane fusion, resulting in cell division arrest at the late telophase. This study provides the first evidence that the plasma membrane phospholipids act as a regulator of the dynamic movement of cytoskeletal proteins and play a crucial role in mediating a coordinate movement between the contractile ring and plasma membrane to achieve successful cell division.
MATERIALS AND METHODS
Materials.
Egg phosphatidylcholine was prepared by chromatography on aluminum oxide neutral and Iatrobeads. Dicetyl phosphate, cholesterol, BSA, and nocodazole were purchased from Sigma. PE, phosphatidylserine, phosphatidylinositol, phosphatidic acid, and cardiolipin were purchased from Avanti Polar Lipids.
Cell Culture.
CHO-K1 fibroblasts (a gift from M. Nishijima, National Institute of Health, Tokyo) were grown in Ham’s F12 medium containing 10% newborn calf serum at 37°C in a 5% CO2/95% air incubator. Mitotic CHO-K1 cell populations were isolated from monolayer cultures grown in 850-cm2 plastic roller bottles (Falcon) as described (14). Briefly, each bottle was seeded with 8.8 × 106 cells in 100 ml growth medium. After incubation at 37°C for 48 h, the culture medium was replaced with growth medium containing 0.04 μg/ml nocodazole. The cells were incubated for further 2 h at 37°C, and prometaphase cells were isolated by shaking.
Binding of Ro-SA Complex to Model Membranes.
Ro was biotinylated as described (17). 125I-labeled (125I)-SA was conjugated with biotinylated Ro and separated from free biotinylated Ro using a PD-10 gel filtration column (Pharmacia LKB). Multilamellar liposomes were prepared by vortex mixing, incubated with 125I-SA-Ro (50,000 cpm) in TBS, pH 7.4, containing 1% (wt/vol) BSA for 30 min at room temperature, after which the reaction mixture was centrifuged at 20,000 × g for 5 min. The resulting pellet was washed three times with TBS containing 1% (wt/vol) BSA and the radioactivity of the precipitated liposomes was counted using a γ-counter.
Binding of SA-Ro Complex to Erythrocyte Membranes.
Fluorescein (FL)-SA (Vector Laboratories) was conjugated with biotinylated Ro and purified by gel filtration as described above. Human blood, freshly drawn from healthy volunteers, was heparinized and centrifuged at 300 × g for 5 min at room temperature to obtain the erythrocytes, which were washed three times with 10 vol of ice-cold PBS. Streptolysin O (SLO; kindly provided by S. Bhakdi, University of Mainz, Germany)-permeabilized erythrocytes were prepared by incubating 5 × 107 cells in 0.5 ml of 1 μg/ml SLO in incubation buffer (25 mM Hepes, pH 7.4/115 mM potassium acetate/2.5 mM MgCl2/1 mM DTT) for 30 min at room temperature and washed three times with the incubation buffer (18). Under these conditions, 100% of the hemoglobin was released by the erythrocytes. Freshly prepared or SLO-permeabilized erythrocytes (5 × 107 cells) were incubated with either 5 μg/ml FL-SA-Ro or 125I-SA-Ro (20,000 cpm) in Hanks’-buffered saline (HBS) containing 0.5% BSA (0.5% BSA-HBS) for 30 min on ice. After the incubation, the erythrocytes were washed three times with cold 0.5% BSA-HBS and permeabilized with 1 μg/ml SLO to remove the hemoglobin. The labeled SLO-permeabilized erythrocytes were washed three times with cold 0.5% BSA-HBS and then subjected to fluorescence microscopy or γ-counting. In some experiments, either FL-SA-Ro or 125I-SA-Ro was preincubated with 100 μM PE-containing liposome and the mixture was used for the binding assay. Fluorescence microscopy was performed using a Zeiss Axioplan microscope equipped with Planneofluar 100× objective.
Binding of FL-SA-Ro to Dividing CHO Cells.
Prometaphase cells (2 × 105) were prepared as described above. The cells were washed with 0.5% BSA-HBS to remove nocodazole and incubated at 37°C for various period and labeled with 0.2 μg/ml 4′-6-diamino-2-phenylindole at 37°C for 10 min in 0.5 ml of 0.5% BSA-HBS. Then the cells were incubated with 5 μg/ml FL-SA-Ro at 4°C for 30 min, washed with 0.5% BSA-HBS and photographed. In some experiments, after incubation with FL-SA-Ro, the cells were fixed with 3.7% (wt/vol) formaldehyde in PBS at 25°C for 20 min, washed and incubated with 40 ng/ml tetramethylrhodamine isothiocyanate (TRITC)-phalloidin at 25°C for 1 h. The cells were washed and photographed.
Cell Division Arrest by SA-Ro.
Prometaphase cells were incubated with various concentrations of either SA, SA-Ro, or 7.2 μM SA-Ro preabsorbed with 100 μM PE-containing liposomes. The effects of SA-Ro on cell division were quantified by counting the number of the cells in dumbbell-shaped cytokinetic form per field of view, one field containing ≈100-150 cells. In one experiment, the effects were quantified by determining the percentage of the cells that were in the cytokinetic form in the photographic field and calculating the average percentage of 10 different photographs. The data shown in this paper are averages of three independent experiments.
Localization of F-Actin and Microtubules in SA-Ro-Arrested Cells.
SA-Ro-treated cells were fixed with 3.7% (wt/vol) formaldehyde in PBS, washed, and blocked with PBS containing 10% (wt/vol) goat serum for 30 min at 25°C. The cells were incubated with rat antiyeast tubulin monoclonal (ICN) antibodies diluted with PBS containing 10% goat serum to 1/100 for 16 h at 4°C. Then the cells were washed with PBS and incubated with rhodamine-conjugated goat anti-rat IgG (Cappel) diluted to 1/200 with PBS containing 10% goat serum for 1 h at 37°C. For F-actin staining, fixed cells were incubated with TRITC-phalloidin as described above. Fluorescence microscopy was performed using a Zeiss Axioplan microscopy and the fluorescent signals were recorded as gray-scale digital image using Hamamatsu Photonics (Hamamatsu, Japan) system. Images were then converted to Photoshop format (Adobe Systems, Mountain View, CA).
RESULTS AND DISCUSSION
Localization of PE on Biological Membranes.
Ro is a 19-amino acid tetracyclic polypeptide isolated from S. griseoverticillatum, which forms a tight equimolar complex with PE on biological membranes (15–17). We conjugated Ro with SA, which was labeled with fluorescein or 125I. Although the free Ro molecules are cytotoxic, conjugation of the biotinylated Ro with SA abolished their cytotoxicity (data not shown). The binding of the SA-Ro complex to PE was confirmed to be specific by studying its binding to liposomes containing various phospholipids (Fig. 1A).
Figure 1.
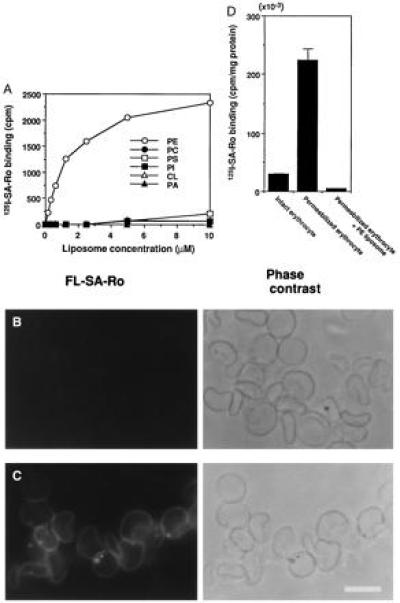
Specific binding of SA-Ro complex to PE. (A) Ro conjugated with 125I-SA-Ro bound selectively to PE-containing liposomes. 125I-SA-Ro was incubated with various concentrations of liposomes composed of egg yolk phosphatidylcholine (PC), dicetyl phosphate and cholesterol (respective molar ratios, 0.75/0.1/1.0) without (•) or with PE (○), phosphatidylserine (PS, □), phosphatidylinositol (PI, ▪), phosphatidic acid (PA, ▴), or cardiolipin (CL, ▵) each at a molar ratio to egg yolk phosphatidylcholine of 0.25. (B and C) Ro conjugated with FL-SA stained permeabilized erythrocyte membranes, but not intact erythrocytes. FL-SA-Ro was added to intact (B) or SLO-treated erythrocytes (C) at 4°C. The intact erythrocytes were permeabilized with SLO after FL-SA-Ro treatment and both the samples were washed and photographed. FL-SA-Ro fluorescence and phase contrast of same specimens, respectively, are shown. Intact erythrocytes were virtually unstained, whereas permeabilized membranes were labeled uniformly. (D) Binding of 125I-SA-Ro to intact and permeabilized erythrocytes. Permeabilized erythrocytes bound 10 times more 125I-SA-Ro than intact erythrocytes and preadsorption of 125I-SA-Ro with PE-containing liposomes abolished this binding (n = 3). (Bar = 10 μm.)
It is well-established that phospholipids in biological membranes are distributed asymmetrically between the inner and outer leaflet of the lipid bilayer (7). In human erythrocyte membranes, most of the PE molecules are located on the inner leaflet of the membrane bilayers and this asymmetric distribution of PE is also observed in various eukaryotic cell membranes (12, 13). To assess whether Ro coupled with FL-SA-Ro could detect PE in biomembranes, we examined the binding of FL-SA-Ro to intact and permeabilized erythrocyte membranes. FL-SA-Ro did not stain intact erythrocytes appreciably (Fig. 1B), and treating erythrocytes with 0.25% (wt/vol) trypsin did not change the FL-SA-Ro binding, suggesting that it was not inhibited by cell surface proteins (data not shown). In contrast, when FL-SA-Ro was added to SLO-permeabilized erythrocytes (18), the entire erythrocyte membranes were labeled strongly (Fig. 1C), whereas no significant staining was observed when FL-SA-Ro was preabsorbed with PE-containing liposome. SLO-permeabilized erythrocyte membranes bound ≈10× more 125I-SA-Ro than intact erythrocytes and preadsorption of 125I-SA-Ro with the PE-containing liposome abolished this binding (Fig. 1D). These results are consistent with previous observations that PE is distributed asymmetrically in erythrocyte membranes (8, 9).
Cell Surface Distribution of PE in Dividing CHO-K1 Cells.
Next we examined the cell surface distribution of PE in dividing CHO-K1 cells. During the course of cytokinesis, an assembly of microfilaments called the contractile ring appears just beneath the plasma membrane around the equator of the parent cells (1). The sliding of actin and myosin filaments generate a force that pulls the plasma membrane inward to create cleavage furrow, which narrows to finally pinch the cell in two. FL-SA-Ro brightly labeled the cleavage furrow at late telophase (Fig. 2C), where contractile ring was costained with TRITC-phalloidin (Fig. 2E). Only a few faint dots in cells in prometaphase (Fig. 2A), anaphase (Fig. 2B), and the G1 phase (Fig. 2D), and no significant staining of cells in metaphase and interphase (data not shown) was observed. Some of the cells at the late telophase form the midbody, which remains as a tether between the two daughter cells. Among 36 cells identified to be at the late telophase in the five independent experiments, none of the 14 cells with the midbody formation showed any significant staining, whereas the remaining 22 cells with the similar morphological feature to those shown in Fig. 2C showed the dramatic staining at the cleavage furrow (data not shown). Neither FL-SA without Ro nor FL-SA-Ro preincubated with PE-liposomes showed any staining and the cell-cell contact regions between the cells at other stages were not stained (data not shown). We also confirmed by blotting analyses that SA-Ro complex showed no crossreactions with the cellular proteins (data not shown). These results indicate that PE was exposed on the cell surface at the cleavage furrow during the specific period of the late telophase, just prior to the midbody formation.
Figure 2.

FL-SA-Ro selectively labeled the cleavage furrows of intact CHO cells during cytokinesis. Cells at prometaphase (A), anaphase (B), late telophase (C), and G1 phase (D) were incubated with FL-SA-Ro for 30 min at 4°C, then washed and photographed. Phase contrast micrographs, FL-SA-Ro staining and 4′-6-diamino-2-phenylindole (DAPI) fluorescence of the same specimens, respectively, are shown. (E) Late telophase cells were double-labeled with FL-SA-Ro and TRITC-phalloidin. Note that strong fluorescence was observed only at the cleavage furrow during cytokinesis; indicated by arrows in C. (Bar = 10 mm.)
There are two possible explanations for this specific staining of the cleavage furrow by FL-SA-Ro: (i) that PE was concentrated at the cleavage furrow due to lateral movement on the cell surface as observed with various cell surface and cytoskeletal components (19–21), and (ii) that PE was newly exposed on the cell surface as a result of scrambling of the phospholipids during late telophase of cytokinesis. The following observations support the latter hypothesis. First, a previous study (14) showed that the amount of PE in the outer leaflets of CHO cell plasma membranes increased about 3-fold when the cells entered the late telophase/G1 phase. Second, in this study, intense staining was observed only during late telophase and we detected no alterations in the surface distribution of FL-SA-Ro during cytokinesis (data not shown).
Cell Division Arrest by SA-Ro.
To explore the role of PE exposed on the cell surface during cell division, we examined the effect of SA-Ro on cytokinetic progression. We found that adding SA-Ro to prometaphase cells followed by incubation for 2 h blocked cytokinesis at late telophase (Fig. 3B). The effect of SA-Ro was concentration-dependent and cytokinesis of about 80% of the cells was inhibited after incubation with 7.2 μM SA-Ro (Fig. 3C). Neither SA nor SA-Ro preincubated with PE had any effects on cytokinesis (Fig. 3 A and C). Upon interacting with PE, Ro was demonstrated to form a firm complex with PE, which was not dissociated by treatment with organic solvents, such as 1-butanol (15–17). The previous study (17) showed that Ro trapped the cell surface PE exposed onto the cell surface, suggesting that surface trapping of PE arrested cytokinesis.
Figure 3.

Blockade of cell division by SA-Ro. SA (A) or SA-Ro (B), both 7.2 μM, was added to prometaphase cells, and then the cells were incubated at 37°C for 2 h, washed, and photographed. Cells in telophase/late telophase accumulated after SA-Ro treatment. (Bar = 30 μm.) (C) Concentration-dependent cell division arrest by SA-Ro. SA-Ro, ○; SA without Ro, •; 7.2 μM SA-Ro preabsorbed with PE-containing liposomes, (▵).
SA-Ro treatment affected neither actomyosin contraction, microtubular rearrangement nor nuclear division, but arrested cytokinesis specifically at late telophase. Forty-five minutes after adding SA-Ro to prometaphase cells, furrowing and chromosome separation proceeded normally and both the arrays of microtubules and contractile ring formed by F-actin were clearly seen between the poles of two daughter cells (Fig. 4 A and B). Two hours after the addition of SA-Ro, the arrested cells showed typical features of cells in G1 phase where microtubules showed a radial array (Fig. 4D), while F-actin was still clearly seen as a ring in the midregions of the two daughter cells (Fig. 4C). Light and electron microscopic analyses have shown that neither midbody formation nor membrane fusion was occurred in the arrested cells (data not shown). The contractile ring is shown to undergo an isovolumetric contraction during the early stages of furrowing and then disassemble during the completion of cell cleavage (1). Reagents that affect microtubular assembly, such as nocodazole and taxol, inhibited cortical movement and actin organization. Cytochalasin, which inhibits actin assembly, blocked actomyosin contraction, resulting in arrest of cytokinesis at the early stages of furrowing (1, 22, 23). SA-Ro is a unique reagent that specifically inhibits the contractile ring disassembly and subsequent membrane fusion, causing cell division arrest at late telophase. It is likely that cell surface immobilization of PE caused the disorganized movement of the cytoskeletal proteins, resulting in the inhibition of the subsequent cytokinetic process, plasma membrane fusion.
Figure 4.
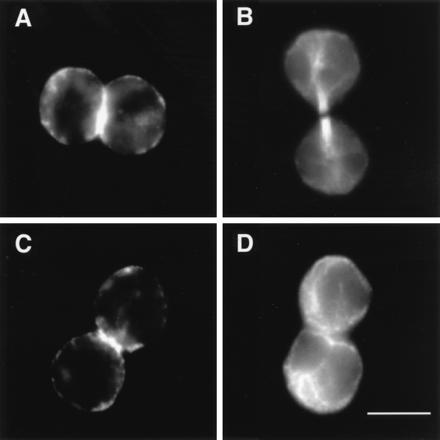
Localization of F-actin and microtubules in SA-Ro-arrested cells. Prometaphase cells were incubated for either 45 min (A and B) or 120 min (C and D) in the presence of SA-Ro, and the SA-Ro-treated cells were fixed and stained with TRITC-phalloidin (A and C) or rat antiyeast tubulin monoclonal antibody (B and D). (Bar = 10 μm.)
Several studies have shown that certain structural features of the membrane lipid bilayer may play a role in modulating the activity of various proteins (24, 25). In particular, PE tends to form a nonbilayer hexagonal structure that has been shown to activate various membrane-bound enzymes such as protein kinase C, calcium pomp, and phospholipase D (26–29). Recent studies have shown that nonmuscle myosin II binds to phospholipid vesicles via the tail region and that this membrane-myosin II interaction regulates both the assembly and disassembly of myosin filaments (30–32). Experiments with Dictyostelium expression myosin II with the truncated tail region provide evidence of the importance of myosin filament disassembly in the function of the contractile ring, implying that the aberrant organization of PE in the membrane may affect the myosin II function, causing the inhibition of the contractile ring disassembly (33). It is also intriguing that the organization of the membrane phospholipids may affect the function of other proteins, such as the spectrins, band 4.1 superfamily proteins, ankyrins, and annexins, which have been suggested to link between actin cytoskeletons and cellular membranes (19–21, 34).
In conclusion, this study shows that the redistribution of the plasma membrane phospholipids may play a pivotal role in mediating a coordinate movement between the contractile ring and plasma membrane to achieve successful cell division. The fluorescence-labeled peptide developed in this study will provide a useful tool for probing the molecular motion and function of PE in biological membranes.
Acknowledgments
We thank Dr. H. Ishitsuka for providing Ro09-0198. This work was supported by Grants-in-Aid for Science Research from the Ministry of Education and Culture of Japan and Research Fellowships of the Japan Society for the Promotion of Science for Young Scientists.
Footnotes
Abbreviations: Ro, Ro09-0198; SA, streptavidin; SA-Ro, SA conjugated to biotinyl Ro; CHO, Chinese hamster ovary; FL-SA-Ro, fluorescein-labeled SA-Ro; 125I-SA-Ro, 125I-labeled SA-Ro; PE, phosphatidylethanolamine; SLO, streptolysin O; TRITC, tetramethylrhodamine isothiocyanate; HBS, Hanks’ buffered saline.
References
- 1.Schroeder T E. Ann NY Acad Sci. 1990;582:78–87. doi: 10.1111/j.1749-6632.1990.tb21669.x. [DOI] [PubMed] [Google Scholar]
- 2.Satterwhite L L, Pollard T D. Curr Opin Cell Biol. 1992;4:43–52. doi: 10.1016/0955-0674(92)90057-j. [DOI] [PubMed] [Google Scholar]
- 3.Fishkind D J, Wang Y. Curr Opin Cell Biol. 1995;7:23–31. doi: 10.1016/0955-0674(95)80041-7. [DOI] [PubMed] [Google Scholar]
- 4.Zimmerberg J, Vogel S S, Chernomordik L V. Annu Rev Biophys Biomol Struct. 1993;22:433–466. doi: 10.1146/annurev.bb.22.060193.002245. [DOI] [PubMed] [Google Scholar]
- 5.Ellens H, Siegel D P, Alford D, Yeagle P L, Boni L, Lis L J, Quinn P J, Bentz J. Biochemistry. 1989;28:3692–3703. doi: 10.1021/bi00435a011. [DOI] [PubMed] [Google Scholar]
- 6.Verkleij A J, Leunissen-Bijivelt J, De Kruijiff B, Hope M, Culluis P R. CIBA Found Symp. 1984;103:45–59. doi: 10.1002/9780470720844.ch4. [DOI] [PubMed] [Google Scholar]
- 7.Zachowski A. Biochem J. 1993;294:1–14. doi: 10.1042/bj2940001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zwaal R F A, Roelofsen B, Comfurius P, Van Deenen L L M. Biochim Biophys Acta. 1975;406:83–96. doi: 10.1016/0005-2736(75)90044-9. [DOI] [PubMed] [Google Scholar]
- 9.Shroit A J, Zwaal R F A. Biochim Biophys Acta. 1991;1071:313–329. doi: 10.1016/0304-4157(91)90019-s. [DOI] [PubMed] [Google Scholar]
- 10.Sessions A, Horwitz A F. FEBS Lett. 1981;134:75–78. doi: 10.1016/0014-5793(81)80554-6. [DOI] [PubMed] [Google Scholar]
- 11.Santini M T, Indovina P L, Cantafora A, Blotta I. Biochim Biophys Acta. 1990;1023:298–304. doi: 10.1016/0005-2736(90)90426-o. [DOI] [PubMed] [Google Scholar]
- 12.Herrmann A, Clague M J, Puri A, Morris S J, Blumenthal R, Grimaldi S. Biochemistry. 1990;29:4054–4058. doi: 10.1021/bi00469a005. [DOI] [PubMed] [Google Scholar]
- 13.Lucy J A. Biochem Soc Trans. 1993;21:250–253. doi: 10.1042/bst0210280. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi T, Pagano R E. J Biol Chem. 1989;264:5966–5973. [PubMed] [Google Scholar]
- 15.Choung S-Y, Kobayashi T, Inoue J, Takemoto K, Ishitsuka H, Inoue K. Biochim Biophys Acta. 1988;940:171–179. doi: 10.1016/0005-2736(88)90192-7. [DOI] [PubMed] [Google Scholar]
- 16.Wakamatsu K, Choung S-Y, Kobayashi T, Inoue K, Higashijima T, Miyazawa T. Biochemistry. 1990;29:113–118. doi: 10.1021/bi00453a013. [DOI] [PubMed] [Google Scholar]
- 17.Aoki Y, Uenaka T, Aoki J, Umeda M, Inoue K. J Biochem (Tokyo) 1994;116:291–297. doi: 10.1093/oxfordjournals.jbchem.a124522. [DOI] [PubMed] [Google Scholar]
- 18.Bhakdi S, Weller U, Walev I, Martin E, Jonas D, Palmer M. Med Microbiol Immunol. 1993;182:167–175. doi: 10.1007/BF00219946. [DOI] [PubMed] [Google Scholar]
- 19.Berlin R D, Oliver J M, Walter R J. Cell. 1978;15:327–341. doi: 10.1016/0092-8674(78)90002-8. [DOI] [PubMed] [Google Scholar]
- 20.Sato N, Yonemura S, Obinata T, Tsukita S, Tsukita S. J Cell Biol. 1991;113:321–330. doi: 10.1083/jcb.113.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yonemura S, Nagafuchi A, Sato N, Tsukita S. J Cell Biol. 1993;120:437–449. doi: 10.1083/jcb.120.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoyt M A, Totis L, Roberts T. Cell. 1991;66:507–517. doi: 10.1016/0092-8674(81)90014-3. [DOI] [PubMed] [Google Scholar]
- 23.Carter S B. Nature (London) 1967;231:261–264. doi: 10.1038/213261a0. [DOI] [PubMed] [Google Scholar]
- 24.Yeagle P L. FASEB J. 1991;3:1833–1842. [PubMed] [Google Scholar]
- 25.Igarashi K, Kaneda M, Yamaji A, Saido T C, Kikkawa U, Ono Y, Inoue K, Umeda M. J Biol Chem. 1995;270:29075–29078. doi: 10.1074/jbc.270.49.29075. [DOI] [PubMed] [Google Scholar]
- 26.Navarro J, Toivio-Kinnucan M, Racker E. Biochemistry. 1984;23:130–135. doi: 10.1021/bi00296a021. [DOI] [PubMed] [Google Scholar]
- 27.Yeagle P L, Sen A. Biochemistry. 1986;25:7518–7522. doi: 10.1021/bi00371a039. [DOI] [PubMed] [Google Scholar]
- 28.Bazzi M D, Youakin M A, Nelsestuen G L. Biochemistry. 1992;31:1125–1134. doi: 10.1021/bi00119a022. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura S, Kiyohara Y, Jinnai H, Hitomi T, Ogino C, Yoshida K, Nishizuka Y. Proc Natl Acad Sci USA. 1996;93:4300–4304. doi: 10.1073/pnas.93.9.4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murakami N, Elzinga M, Singh S S, Chauhan V P S. J Biol Chem. 1994;269:16082–16090. [PubMed] [Google Scholar]
- 31.Deqin L, Miller M, Chantler P D. Proc Natl Acad Sci USA. 1994;91:853–857. doi: 10.1073/pnas.91.3.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murakami N, Singh S S, Chauhan V P S, Elzinga M. Biochemistry. 1995;34:16046–16055. doi: 10.1021/bi00049a019. [DOI] [PubMed] [Google Scholar]
- 33.Egelhoff T T, Brown S S, Spudich J A. J Cell Biol. 1991;112:677–688. doi: 10.1083/jcb.112.4.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schroer T A, Bingham J B, Gill S R. Trends Cell Biol. 1996;6:212–215. doi: 10.1016/0962-8924(96)20014-5. [DOI] [PubMed] [Google Scholar]