

Abstract
The primase DnaG of Escherichia coli requires the participation of the replicative helicase DnaB for optimal synthesis of primer RNA for lagging strand replication. However, previous studies had not determined whether the activation of the primase or its loading on the template was accomplished by a helicase-mediated structural alteration of the single-stranded DNA or by a direct physical interaction between the DnaB and the DnaG proteins. In this paper we present evidence supporting direct interaction between the two proteins. We have mapped the surfaces of interaction on both DnaG and DnaB and show further that mutations that reduce the physical interaction also cause a significant reduction in primer synthesis. Thus, the physical interaction reported here appears to be physiologically significant.
Keywords: protein–protein interaction, lagging strand replication
The multiprotein complex that initiates, elongates, and terminates DNA replication has been likened to a protein machine (1). The various component proteins of this machine have to work coordinately to successfully carry out the complex task of DNA replication. Thus, careful analyses of the interactions amongst the replisomal proteins are of critical importance for gaining further insights into the mechanism and control of the three steps of DNA replication. In both Escherichia coli and in mammalian cells, discontinuous synthesis of Okazaki fragments is primed by a class of enzymes called primases (2) that move along the length of the DNA template as a part of a multiprotein primosomal complex, synthesizing RNA primers at intermittent locations on the template DNA (3). Although a great deal of information has been uncovered regarding the enzymology of fork movement in prokaryotes and eukaryotes (3, 4), a number of significant questions still remain unanswered regarding the protein–protein interactions that control and drive fork movement.
Studies on priming of phage single-stranded DNA (ssDNA), which is not coated with ssDNA binding protein, have shown that DnaB helicase (5) is needed along with DnaG primase for optimal primer synthesis (3). Two alternative mechanisms have been proposed to account for the need for DnaB in primer synthesis. One hypothesis was that DnaB interacted with ssDNA and created a conformation that allowed DnaG to load on to the DNA. This idea was based on the observation that DnaB binding induced a change in the secondary structure of ssDNA (5). The alternative hypothesis was that DnaB interacted directly with DnaG and with ssDNA, thus facilitating the loading of the primase on the template and/or the activation of the enzyme (3). Although functional interaction between the C terminus of DnaG with DnaB was reported by Marians and coworkers (7, 8), critical evidence showing physical interaction between the two protein has been elusive.
To distinguish between the two alternative models of activation of the primase and/or its loading on DNA, we have investigated the possible physical interaction between the helicase and the primase of Escherichia coli. In this paper we present evidence supporting direct protein–protein interaction in vitro between the two enzymes. We have localized the interacting surfaces on both enzymes and have isolated mutations in DnaB that reduce the protein–protein interaction without causing a significant reduction in helicase activity. We show further that a mutant form of DnaB that is impaired in DnaB–DnaG interaction, also suffered from an ≈4- to 5-fold reduction in promoting primer synthesis in vitro in comparison with wild-type (wt) DnaB. Conversely, we have shown that a mutant form of DnaG that is defective in DnaB-stimulated synthesis of primer RNA also showed reduced physical interaction with wt DnaB. Thus, direct physical interaction between the primase and the helicase appears to be necessary for optimal primer synthesis.
MATERIALS AND METHODS
Plasmids and Bacterial Strains.
The E. coli strains JM109 (sup E44, rel A1, rec A, end A1, gyr A96, hsd R17D, Δ [lac, pro AB], [F′tra D36, lacIq, Δ(lac Z)M15, pro A+B+),(rk−,mk+), and BL21[DE3] [F−omp T,hsd S, (rB−,mB−) gal] containing the plasmid pLysS (9) were used for all cloning and protein expression, respectively. The proteins were expressed in the T7 promoter containing vector pET11b (Novagen) or the tac promoter containing pGEX vector (Pharmacia). The pET3c-DnaG Q576A overproducer clone was a gift from Ken Marians (Sloan–Kettering Cancer Center, New York).
Site-Directed Mutagenesis.
Site-directed mutagenesis was performed as described (10). The regions of DnaB having two or more contiguous charged residues were regarded as potential interaction surfaces for DnaG and these sites were replaced by alanine. Specifically, the following charged residues of DnaB were replaced with alanine: mutant m1, D212A and D213A; m2, K216A and K217A; m3, D253A and K254A.
Purification of Proteins and Enzymes.
Wt and mutant forms of DnaG and DnaB were purified as described before (7, 11). The reading frame of full-length DnaG or the N-terminal (bases 1–1265) and the C-terminal domains were fused in frame with the DNA encoding glutathione S-transferase (GST) present in the pGEX vector and the fusion proteins were purified by affinity chromatography on glutathione-agarose. The C-terminal bases 1266–1740 were fused in frame with the His-6 tag present in the vector pET30a (Novagen) and the fusion protein was purified on Ni-nitrilotriacetic acid-agarose columns.
ELISA.
The procedure used was similar to the one published in ref. 12. One member of interacting pair of proteins was dissolved in 100 ml of coating buffer (0.1 M NaHCO3, pH 9.2) at a concentration of 100 μg/ml and adsorbed to each well of a 96-well plastic microtiter plate (Corning) by incubating for 1 hr at 20°C. The wells were rinsed with 200 μl of wash buffer (20 mM Tris, pH 7.4/0.15 M NaCl/0.5% Tween 20). Blocking buffer (200 μl of 5% BSA/0.5% Tween 20 in wash buffer) was added to each well and incubated for 1 hr at 20°C. The adsorbed protein in each well was challenged with 10–500 ng of the second protein dissolved in 100 μl of the blocking buffer and incubated at 20°C for 1 hr. Each well was washed five times with 200 μl of wash buffer. Rabbit antiserum raised against the second protein was diluted 2000-fold in blocking buffer, and 100 μl of the diluted antibody was added to the designated wells and incubated for 1 hr at 20°C. After five washes with the washing buffer, a 1:40,000-fold dilution of a goat anti-rabbit IgG antiserum that was conjugated with alkaline phosphatase (Sigma) was added to each of the wells and incubated further for 1 hr at 20°C. Following five washes with the wash buffer, 100 μl of the chromogenic substrate, p-nitrophenyl disodium phosphate hexahydrate was added to each well and incubated at 20°C for 5–20 min. The absorbance was read at 405 nm in an ELISA plate reader.
Coupled in Vitro Transcription-Translation.
DNAs encoding full-length wt DnaB, mutant forms of DnaB, or peptides of DnaB were cloned downstream from a T7 promoter and transcribed and translated in vitro in the presence of 35S-methionine (10 mCi/ml; 1 Ci = 37 GBq), 10 mM amino acid mixture, four units of T7 RNA polymerase, and 40 units of an RNase inhibitor in a rabbit reticulocyte lysate system (Promega) at 30°C for 2 hr to generate labeled peptides. Luciferase was similarly labeled and used as a control. The labeled products were analyzed by SDS/PAGE followed by autoradiography.
Affinity Chromatography.
GST and GST-fusion proteins (2.5–10 μg) in 50 μl of TBS buffer (150 mM NaCl/20 mM Tris·Cl, pH 7.2) were incubated with 40 μl of packed volume of glutathione-agarose beads and incubated on ice for 45 min. The beads were blocked in 5% BSA in TBS buffer for an additional 1 hr on ice. The beads were recovered by sedimentation. Various amounts of the labeled proteins in blocking buffer supplemented with 5 mM MgCl2/1 mM ATP were added to the pelleted beads and incubated on ice for 30 min. The beads were then washed with 300 μl of 0.5% Tween 20 in TBS and the supernatant was recovered to measure the amount of unbound protein. The beads were extracted with 0.1% SDS, 10 mM 2-mercaptoethanol, 10 mM Tris·Cl (pH 7.4). The eluted samples were analyzed by SDS/PAGE followed by autoradiography.
Helicase Assays.
These were carried out as described (11).
In Vitro Synthesis of Primers.
The reaction mixture contained in 25 μl: 50 mM Tris·Cl (pH 7.6), 5 mM MgCl2, 4 mM DTT; 250 μg/ml BSA, 100 mM of dGTP, dTTP, and dCTP, 100 mM [α-32P]dATP (2000 cpm/pmol), 100 mM each of GTP, CTP, and UTP, 1 mM ATP, 0.1 μg of rifampicin, 1 unit of RNA guard (Pharmacia), 1 unit of Klenow fragment of DNA polymerase I, 200 ng of M13 ssDNA, and various amounts of wt or mutant forms of DnaB and wt DnaG. The reaction mixtures were incubated for 30 min at 30°C. The reactions were stopped by adding EDTA (pH 8.0) to 200 mM, spotted on DE81 (Whatman) filter paper discs. The discs were washed three times with 5 ml of 10% cold trichloroacetic acid containing 10 mM pyrophosphate and twice with 95% ethanol, dried, and counted. The addition of DNA polymerase (Klenow fragment) helped to amplify the signal by extending the primers synthesized by DnaG on template ssDNA (14).
RESULTS
DnaB and DnaG Physically Interact in Vitro with Each Other.
Because evidence supporting direct interaction between the two proteins had been elusive, we assumed that such interaction, if it occurred, would probably be weak. We therefore endeavored to modify and develop procedures to detect this potentially weak interactions. We modified ELISA to detect the possible interaction between the helicase and the primase as described in a previous section. The modifications were in the washing procedure to reduce the background nonspecific binding. Different amounts of DnaG were adsorbed onto the wells of plastic microtiter plates and blocked the remainder of the free surface with BSA. We challenged the immobilized DnaG with various amounts of purified DnaB. DnaA protein, which is known to interact with DnaB, was also immobilized on plastic and used as a positive control, whereas helicase II was used as a negative control. The binding of DnaB to DnaG, DnaA, and helicase II was measured as described in a preceding section. The results showed that DnaB readily bound to the immobilized DnaG. DnaB also bound to DnaA in agreement with previously published data (14). Helicase II did not show significant levels of interaction with DnaB (Fig. 1A). We performed reciprocal binding experiments in which purified DnaB, DnaC, and BSA were immobilized on plastic surface and challenged with increasing amounts of DnaG. The results showed that DnaG bound to immobilized DnaB but no significant binding to immobilized DnaC and BSA was detectable (Fig. 1B).
Figure 1.
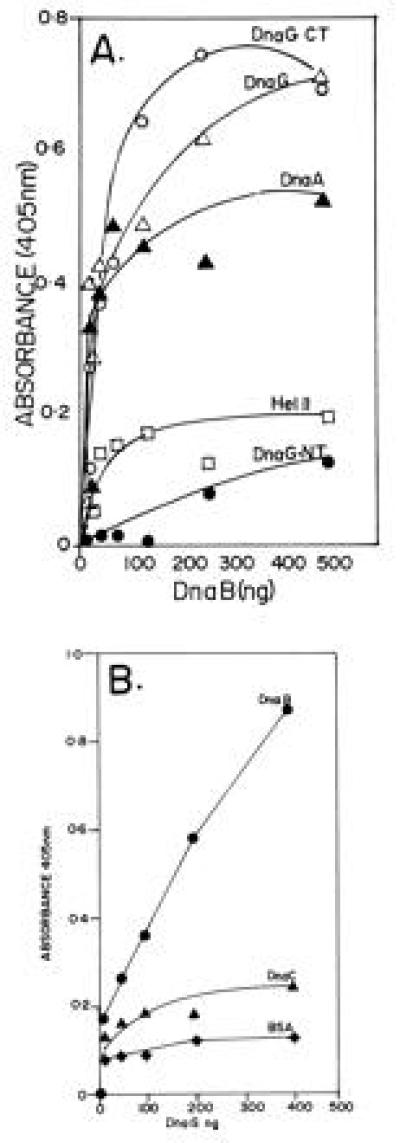
ELISA showing the interaction of DnaG primase with DnaB helicase. (A) Interaction between immobilized DnaG with DnaB in solution. Full-length DnaG and its N-terminal (DnaG-NT) and C-terminal (DnaG-CT) peptides, DnaA, and helicase II (Hel II) were immobilized on plastic surfaces of the wells of microtiter plates and were challenged with various amounts of DnaB in solution. Note that full-length DnaG and its C-terminal peptide show clear binding to DnaB. The DnaA protein, used as a positive control also binds, as expected, to DnaB. In contrast the N-terminal peptide of DnaG and helicase II (negative control) elicited low or background levels of binding signal. (B) Reciprocal binding of immobilized DnaB to DnaG in solution. DnaB, DnaC (negative control), and BSA (negative control) were immobilized on the plastic surface of microtiter plates and challenged with various amounts of DnaG in solution. Whereas DnaG readily bound to immobilized DnaB, there was only low levels of binding of DnaG to immobilized DnaC and BSA.
We wished to confirm the observed DnaB–DnaG interaction by an independent technique. We prepared 35S-labeled DnaB by coupled in vitro transcription-translation. Labeled luciferase was similarly prepared and used as a negative control. We constructed an in-frame, N-terminal fusion of DNA encoding GST with that encoding DnaG. The fusion retained the entire reading frame of DnaG including the ATG codon. The fusion protein was purified and adsorbed to GST-glutathione-agarose beads. Control beads contained GST by itself. An equimolar mixture of labeled luciferase and DnaB was loaded onto glutathione-agarose and DnaG-GST-agarose beads, washed, eluted, resolved by SDS/PAGE, and detected by autoradiography. The autoradiograms showed that DnaB was specifically retained on the DnaG-GST matrix but not on the control GST matrix. Luciferase did not bind to DnaG-GST (or GST) beads and was recovered in the flow through fractions (Fig. 2A). Thus, the affinity column chromatography confirmed the ELISA data showing that DnaG specifically interacted with DnaB helicase.
Figure 2.
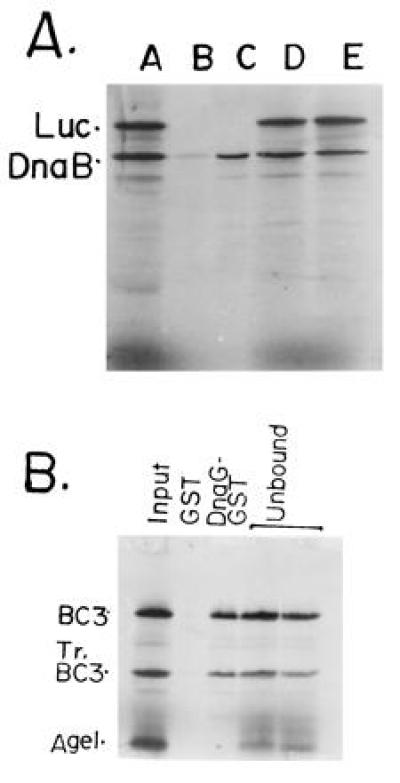
GST-affinity chromatography showing the binding of full-length DnaB and its peptides with DnaG-GST fusion protein affinity matrix. (A) Full-length DnaB and luciferase (Luc; negative control) were labeled with 35S-methionine by coupled in vitro transcription-translation in rabbit reticulocyte extracts. An approximately equimolar mixture of the two labeled proteins were loaded on to the control GST-glutathione-agarose and the DnaG-GST-glutathione-agarose matrices. The flow through fraction was collected and analyzed. The beads were washed and the proteins eluted and resolved by SDS/PAGE. An autoradiogram of the gel shows: lane 1, input mixture of full-length DnaB, a truncated product of DnaB (N-terminal fragment) and full-length luciferase (Luc); lane 2, protein bound to the control GST-agarose matrix; lane 3, protein bound to the DnaG-GST-agarose matrix. Note that full-length DnaB, its truncated peptide, that was generated during the labeling process, were selectively retained on the DnaG-GST matrix but not on the control GST-matrix. No luciferase bound to the control or to the DnaG-GST matrices. (B) DNA template encoding the BC3 and AgeI (see Fig. 3) peptides of DnaB were labeled in vitro as described. The In vitro labeling process generated the tr.BC3 (Tr.BC3) peptide. The truncation must have generated a N-terminal peptide because of the upstream location of the T7 promoter used to transcribe the DNA. Lanes: 1, input mixture of BC3, TrBC3 and the AgeI peptides; 2, binding to the control GST matrix; 3, binding to the DnaG-GST matrix; 4, flow through from the GST matrix; 5, flow through from the DnaG-GST matrix. Note that the BC3 and tr.BC3 peptides (≈40–50% of the input) were retained on the DnaG-GST but not on the control GST matrix. The AgeI peptide was not retained by either matrices and was recovered in the flow through fractions.
Mapping of the Interaction Surfaces.
To localize the interaction surfaces on DnaB, various N-terminal peptides of DnaB called AgeI, truncated BC3 (tr.BC3), and BC3 were prepared by coupled in vitro transcription-translation of fragments of DNA encoding DnaB. The DNA fragments had an upstream T7 promoter and a ribosome binding site. The labeled peptides were mixed in equimolar amounts and adsorbed to and eluted from control GST-agarose and DnaG-GST agarose matrices. Autoradiograms of SDS/PAGE showed that the BC3 and tr.BC3 peptides were retained on the DnaG-GST matrix but not on the GST matrix. The AgeI peptide was not retained on the DnaG-GST matrix and was recovered in the flow through fraction. Thus the interaction surface of DnaG on DnaB is located in the region bracketed by the AgeI and tr.BC3 sites (Figs. 2B and 3). We have purified the AgeI peptide and have observed by ELISA that it does not interact with DnaG thus confirming the affinity binding data (not shown).
Figure 3.
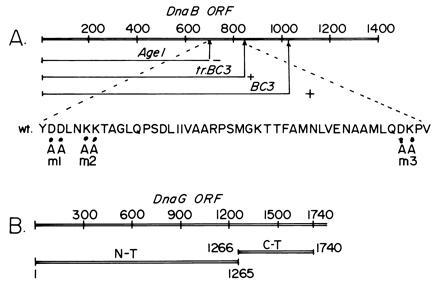
Maps showing the interaction surfaces that participated in the DnaB–DnaG interaction. (A) Map showing DnaB sequence encoding the AgeI, tr.BC3, and BC3 peptides. The region between the AgeI and tr.BC3 binds to DnaB. The amino acid sequence showing the locations of the m1, m2, and m3 mutants that were used to further narrow down the interaction region. (B) The sequence encoding DnaG showing the locations of 1265-bp-long DNA encoding the N-terminal peptide that did not bind to DnaB and the 475-bp-long sequence encoding the C-terminal peptide that bound to DnaB. The numbers refer to base pair coordinates.
We have also mapped the surface of DnaG that interacts with DnaB by separately expressing the N-terminal and C-terminal regions of the primase, purifying the peptides, and performing ELISA with DnaB. The experiments showed that the C-terminal but not the N-terminal peptide of DnaG interacted with DnaB (Fig. 1A). Marians and coworkers (7, 8) had inferred that functional interaction between DnaB and DnaG required the C terminus of the latter protein. Our results therefore are consistent with their observation.
Physiological Relevance of Helicase-Primase Interaction.
Is the physical interaction reported here physiologically significant? We wished to investigate the possible physiological relevance by isolating point mutations in the region of DnaB that interacted physically with DnaG. Three double mutants called m1, m2, and m3, which involved replacement of adjacent charged residues by alanine, were isolated by site-directed mutagenesis (Fig. 3A). Binding studies by ELISA (Fig. 4) and GST matrix affinity binding (data not shown) revealed that m1 and m2 showed noticeable reductions in binding to DnaG. The binding of m3 was very close in magnitude to that of wt DnaB.
Figure 4.
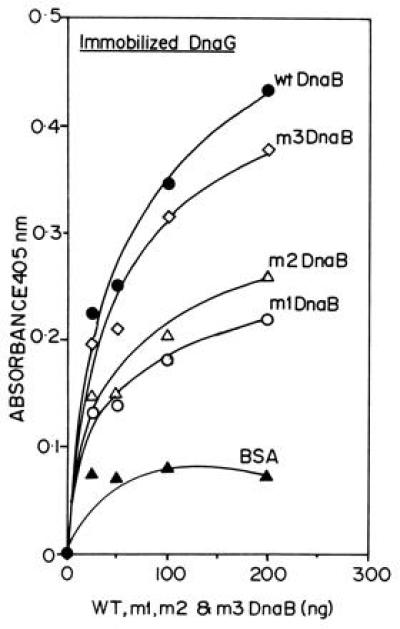
ELISA showing the binding of the m1, m2, and m3 mutant forms of DnaB with immobilized DnaG protein. The data are the averages of three separate experiments. The extent of binding is in the following decreasing order: wt DnaB > m3 > m2 > m1. BSA was used as a negative control.
We expressed and purified the three mutant forms of DnaB and performed helicase assays. The results showed only a small reduction in helicase activity of m1, m2, and m3 proteins in comparison with wt DnaB (Fig. 5). Taking into account the error bars of the data points, m1 showed a/15% reduction, whereas m2 and m3 showed a <10% reduction in helicase activity in comparison with wt DnaB (Fig. 5). We performed priming experiments using M13 ssDNA (not coated with ssDNA binding protein), and purified primase and the normal and the three mutant forms of DnaB proteins. The primer synthesis signal was amplified by extending the newly synthesized primers with the Klenow fragment of DNA polymerase I in the presence of [α-32P]dATP and the other three unlabeled dNTPs. In four independent experiments, we consistently observed a 4- to 5-fold reduction in priming activity of m1 as compared with wt DnaB. The m2 mutant was more active than m1 but still showed a reduction in activity when compared with the wt helicase. The m3 form did not show a significant reduction in priming activity (Fig. 6).
Figure 5.
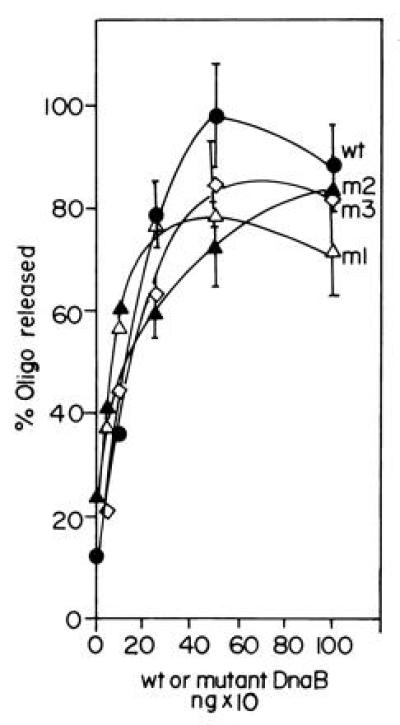
Helicase assay showing the activities of wt and the m1, m2, and m3 forms of DnaB. The mutant forms of DnaB helicase have activities in the range of at least 85–95% of that of the wt enzyme. The helicase assays were performed by hybridizing a labeled 17-mer oligonucleotide to single-stranded M13 DNA and measuring the ATP-dependent unwinding by nondenaturing PAGE. The released radioactive oligodeoxynucleotide bands were excised from the gel and counted.
Figure 6.
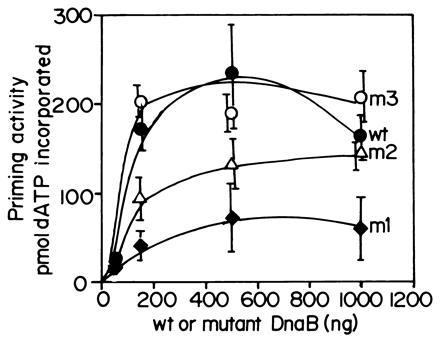
The relative abilities of wt and the mutant forms of DnaB to activate the ability of wt DnaG to synthesize RNA primers on uncoated M13 ssDNA. Note that the m1 mutant shows a 4- to 5-fold reduction in its ability to promote primer synthesis in collaboration with wt DnaG primase. The assay for primer synthesis is described in the text. The m3 mutant does not show detectable reduction in primer synthesis as compared the wt DnaB where as m2 shows more activity than m1 but less than that of the wt helicase.
We have performed the converse experiments using a mutant form of DnaG called Q576A that was isolated by Tougu and Marians (8), to show that the mutant interacts with wt DnaB less strongly than wt DnaG. The DnaG-Q576A mutation, located at the C-terminal region of the reading frame, while retaining normal priming activity in a M13 Gori assay (priming and initiation from the G4 ori in the single-strand to double-strand stage requires DnaG and ssDNA binding protein but no DnaB), showed a 20-fold reduction in DnaB stimulated general priming assay. We purified the Q576A mutant form of DnaG and immobilized it on plastic surface and measured its ability to bind to wt DnaB. Wt DnaG was similarly immobilized and its binding to wt DnaB was also measured by ELISA. The results showed that the Q576A mutant form of DnaG showed a small but consistent reduction in binding to DnaB in comparison with wt DnaG (Fig. 7A). Since the difference in binding of wt DnaG and the Q576A mutant to DnaB was small, we wanted to make sure whether the difference was real. We confirmed the results by performing reciprocal binding experiments in which a constant amount of DnaB was immobilized on the plastic surface of each well and challenged with various amounts of the wt and the Q576A form of DnaG. The results again showed that the mutant form of DnaG in three independent experiments, bound less strongly to DnaB in comparison with the wt primase (Fig. 7B).
Figure 7.
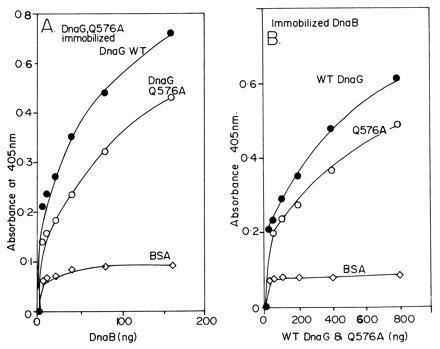
ELISA showing the relative interactions of DnaB with wt DnaG and the mutant form Q576A. (A) Interaction of immobilized wt DnaG and Q576A with wt DnaB in solution. A typical set of results is shown. The wt DnaG consistently bound better than the Q576A protein DnaB in solution. (B) The experiment was performed in converse by immobilizing a constant amount (1 μg per well) of normal DnaB protein and challenging it with equivalent amount of wt and the Q576A protein. The wt DnaG, now present in solution, consistently bound more strongly to the immobilized DnaG than the mutant form.
Taken together, the results showed that direct interaction between the primase and the helicase was necessary for optimal primer synthesis.
DISCUSSION
Keeping in mind the fact that activation and regulation of primer RNA synthesis is an important step in lagging strand DNA synthesis, the results presented in this paper are significant because of unequivocal demonstration of a physical interaction between the DnaB helicase and the DnaG primase of E. coli, identification of the sites of interaction on DnaB and DnaG and for providing the evidence that the interaction was necessary for optimal primer synthesis. Although, there were a number of previous observations reported in the literature that showed functional interaction between the two proteins in phage T4 (15, 16), T7 (17), and E. coli (7, 8), critical evidence showing direct physical interaction between replicative helicase and the primase was, until now, elusive. Marians and coworkers have shown that the functional interaction between DnaB and DnaG involves the C-terminal region of the primase. Our data, showing that the C-terminal but not the N-terminal peptide of DnaG physically interacts with DnaB, are thus consistent with their results (7, 8).
The evidence presented here also maps the surface of DnaB helicase that interacts with DnaG primase, to a small stretch of a maximum of 46 amino acids. The region of the helicase that interacts with plasmid-encoded π initiator protein of R6K has previously been localized to a small region of 37 amino acids immediately upstream of the DnaG interaction site (12). We have purified the N-terminal region of the helicase, that seems to participate in many important protein–protein interactions in a soluble form (unpublished data). The crystallization and determination of the structure of this peptide by x-ray crystallography should provide, in the future, valuable structural information pertaining to the various protein–protein interaction between the helicase and other replication proteins.
We have also mapped the surface of DnaG that physically interacts with DnaB to the C-terminal peptide. On the basis of partial proteolytic digestion of DnaG, Godson and coworkers (18) had shown that the primase had a larger N-terminal domain and a smaller C-terminal domain. The N-terminal domain had the capability to catalyze primer RNA synthesis at the ori of phage G4. However, in the absence of the smaller C-terminal domain, the size of the primer RNA was smaller than normal. The C-terminal domain was therefore believed to have a regulatory function. Similar observations were independently made by Marians and coworkers, who also discovered that the C terminus of DnaG functionally interacts with DnaB helicase. However, critical evidence showing physical interaction between the two enzymes was lacking.
How does the physical interaction between the helicase and the primase enhance the activity of the primase? One could attempt to answer this question by using wt and noninteracting mutant forms of both enzymes to perform kinetic analysis of primer synthesis. Determinations of the crystal structures of both enzymes or at least the important domains should provide valuable information pertaining to the mechanism of primase activation.
The protein–protein interaction between DnaB and DnaG is not exclusively binary as both proteins are known to interact with several other replication proteins. For example, DnaG functionally interacts with the polIII holoenzyme (19). In addition, we have recently discovered that DnaG primase of E. coli physically interacts with the host-encoded DnaA initiator protein and with the plasmid-encoded π and RepA initiator proteins of the plasmids R6K and pSC101, respectively (P.V.A.L.R. and D.B., unpublished data). DnaB is also known to interact with DnaA (14), DnaC (20), the replication terminator proteins of E. coli and Bacillus subtilis (B.K.M., A. Manna, and D.B., unpublished work), and with π initiator protein of R6K and RepA initiator protein of pSC101 (12). It would be interesting to investigate if helicase and primase activities in the replisome are also influenced by interaction with the replication initiator proteins. In fact, as originally suggested by Sueoka and Quinn (21), the origin of replication might be physically associated with the moving replication fork at an anchor point, involving the cell membrane and the initiator protein, thus imparting a looped structure to the replicating chromosome.
Systematic investigations of the various protein–protein interaction taking place in the initiation elongation and termination complexes will undoubtedly continue to provide new insights into the mechanism and control of the three steps of DNA replication.
Acknowledgments
We thank Mr. Mark Lobert for purifying DnaB and DnaG proteins and Dr. Ken Marians for a gift of a mutant strain of DnaG and for informing us about his results prior to publication. This work was supported by the National Institutes of Health Merit Award R37 AI19881 to D.B.
Footnotes
Abbreviations: wt, wild type; GST, glutathione S-transferase, tr.BC3, truncated BC3; ssDNA, single-stranded DNA.
References
- 1.Alberts B M. Trends Genet. 1985;1:26–30. [Google Scholar]
- 2.Rowen L, Kornberg A. J Biol Chem. 1978;253:758–764. [PubMed] [Google Scholar]
- 3.Arai K-I, Kornberg A. Proc Natl Acad Sci USA. 1979;76:4308–4313. doi: 10.1073/pnas.76.9.4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly T J. J Biol Chem. 1988;263:17889–17892. [PubMed] [Google Scholar]
- 5.LeBowitz J, McMacken R. J Biol Chem. 1986;261:4738–4748. [PubMed] [Google Scholar]
- 6.Arai K-I, Kornberg A. J Biol Chem. 1981;256:5260–5266. [PubMed] [Google Scholar]
- 7.Tougu K, Peng H, Marians K J. J Biol Chem. 1994;269:4675–4682. [PubMed] [Google Scholar]
- 8.Tougu K, Marians K J. J Biol Chem. 1996;271:21391–21397. doi: 10.1074/jbc.271.35.21391. [DOI] [PubMed] [Google Scholar]
- 9.Studier F W, Rosenberg A, Dunn J J. Methods Enzymol. 1990;185:60–81. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 10.Kunkel T, Bebeneck K, McClary J. Methods Enzymol. 1991;204:125–138. doi: 10.1016/0076-6879(91)04008-c. [DOI] [PubMed] [Google Scholar]
- 11.Khatri G S, McAllister T, Sista P R, Bastia D. Cell. 1989;59:667–674. doi: 10.1016/0092-8674(89)90012-3. [DOI] [PubMed] [Google Scholar]
- 12.Ratnakar P V A L, Mohanty B K, Lobert M, Bastia D. Proc Natl Acad Sci USA. 1996;93:5522–5526. doi: 10.1073/pnas.93.11.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mendelman L V. Methods Enzymol. 1995;262:405–414. doi: 10.1016/0076-6879(95)62032-x. [DOI] [PubMed] [Google Scholar]
- 14.Marszalek J, Kaguni J. J Biol Chem. 1994;269:4883–4890. [PubMed] [Google Scholar]
- 15.Liu C C, Alberts B M. J Biol Chem. 1981;256:2821–2829. [PubMed] [Google Scholar]
- 16.Venkatesan M, Silver L L, Nossal N G. J Biol Chem. 1982;257:12426–12434. [PubMed] [Google Scholar]
- 17.Scherzinger E, Lanka E, Morelli G, Seifert D, Yuki A. Eur J Biochem. 1977;72:543–558. doi: 10.1111/j.1432-1033.1977.tb11278.x. [DOI] [PubMed] [Google Scholar]
- 18.Sun W, Tormo J, Steitz T A, Godson G N. Proc Natl Acad Sci USA. 1994;91:11462–11466. doi: 10.1073/pnas.91.24.11462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu C A, Zechner E L, Reems J A, McHenry C S, Marians K J. J Biol Chem. 1992;267:4074–4083. [PubMed] [Google Scholar]
- 20.Wickner S, Hurwitz J. Proc Natl Acad Sci USA. 1975;72:921–925. doi: 10.1073/pnas.72.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sueoka N, Quinn W G. Cold Spring Harbor Symp Quant Biol. 1968;33:695–705. doi: 10.1101/sqb.1968.033.01.078. [DOI] [PubMed] [Google Scholar]