

Abstract
The stress-activated protein kinases JNK and p38 mediate increased gene expression and are activated by environmental stresses and proinflammatory cytokines. Using an in vivo model in which oxidative stress is generated in the liver by intracellular metabolism, rapid protein–DNA complex formation on stress-activated AP-1 target genes was observed. Analysis of the induced binding complexes indicates that c-fos, c-jun, and ATF-2 were present, but also two additional jun family members, JunB and JunD. Activation of JNK precedes increased AP-1 DNA binding. Furthermore, JunB was shown to be a substrate for JNK, and phosphorylation requires the N-terminal activation domain. Unexpectedly, p38 activity was found to be constitutively active in the liver and was down-regulated through selective dephosphorylation following oxidative stress. One potential mechanism for p38 dephosphorylation is the rapid stress-induced activation of the phosphatase MKP-1, which has high affinity for phosphorylated p38 as a substrate. These data demonstrate that there are mechanisms for independent regulation of the JNK and p38 mitogen-activated protein kinase signal transduction pathways after metabolic oxidative stress in the liver.
A number of mammalian mitogen-activated protein (MAP) kinases have recently been cloned, including JNK and p38, that have similar activation profiles by environmental stress and proinflammatory cytokines (1–8). These kinases have been shown to phosphorylate and activate a number of transcription factors including, c-Jun, ATF-2, and TCF/Elk-1 (1–4, 7, 9–13). Although there is apparent coordinate regulation of JNK and p38, these protein kinases have distinct substrate specificity; p38 phosphorylates ATF-2 (9) and Elk-1 (13), while JNK phosphorylates c-Jun, ATF-2, and Elk-1 (1–4, 10–12). Similarly, the upstream activating MAP kinase (MAPK) kinases (MKKs) exhibit distinct substrate specificities. MKK3 and MKK6 phosphorylate and activate p38 (13, 14), whereas MKK4 phosphorylates and activates both JNK and p38 (14–16). All extracellular stimulants investigated thus far activate both JNK and p38 MAP kinases. To identify whether p38 and JNK can be independently regulated, we examined a novel model of intracellular stress that is generated by oxidative metabolism in the liver.
In vivo oxidative metabolism of natural metabolites, pharmaceutical drugs, and toxic compounds can lead to oxidative stress in most organs and tissue types. Pharmacological and toxicological studies have demonstrated that the mammalian liver metabolizes various drugs and toxic compounds. Oxidative damage can result through the activities of oxidative enzymes such as P450s and epoxide hydrolase. The oxidative damage is localized to pericentral hepatocytes that are proximal to the central vein of the liver microcirculatory unit. Damage specificity is due to the localization of these oxidative enzymes (17, 18).
We have shown (K.G.M., E. Saez, B. M. Speigelman, and K.E.P., unpublished data) that CCl4-induced oxidative stress results in the activation of AP-1 site-regulated genes, such as c-Jun and glutathione S-transferase (GST) Ya, prior to the appearance of tissue damage. To investigate the mechanisms of AP-1-regulated gene activation by metabolic oxidative stress, we examined AP-1 binding in the liver. An activated AP-1 complex was observed 2–4 hr after injection of CCl4 into mice. Appearance of the activated AP-1 complex was preceded by activation of JNK, which phosphorylates three of the major components of the induced complexes, c-jun, ATF-2, and JunB. Surprisingly, we did not observe activation of p38 MAP kinase, but rather high basal activity in the liver which is rapidly inactivated after CCl4 injection. The mechanism of p38 MAP kinase inactivation was likely coupled to the concomitant transcriptional activation of the phosphatase MKP-1. To our knowledge, these findings demonstrate for the first time that JNK and p38 MAP kinase can be independently regulated.
MATERIALS AND METHODS
C57BL6/J (25 g) mice were given an intraperitonial injection of 100 μl of CCl4 in corn oil, 1:20 dilution. Mice were anesthetized at the time points indicated and livers were perfused with normal saline and removed. Livers (0.5 g) were finely minced and extracted in 5 vol WCE buffer [25 mM Hepes, pH 7.5/0.3 M NaCl/1.5 mM MgCl2/0.2 mM EDTA/0.05% Triton X-100/20 mM β-glycerophosphate/0.1 mM orthovanadate/0.5 mM DTT/0.4 mM phenylmethylsulfonyl fluoride (PMSF)/leupeptin (1 μg/ml)/pepstatin (1 μg/ml)] for 30 min at 4°C. Extracted livers were centrifuged at 100,000 × g at 4°C for 30 min and the cleared lysate was frozen at −80°C.
Electrophoretic mobility-shift assay (EMSA) was performed by incubating 0.5 ng of the c-Jun proximal AP-1 site oligonucleotide labeled with 32P in a 15-μl reaction mixture (20) with 3 μg of liver whole-cell extract for 30 min at room temperature. After incubation for 30 min at room temperature, reaction products were loaded on a 6% polyacrylamide/0.25× TBE gel and electrophoresed at 350 V for 2.5 hr at 4°C. Antibody supershift and immunodepletion were performed by incubating the EMSA binding reaction with 2 μl of antisera to c-fos, c-Jun, ATF-2, JunB, JunD (Santa Cruz Biotechnology), and β-galactosidase (gift of A. S. Yee, Tufts University School of Medicine) at 4°C for 20 min prior to loading on the gel.
The bacterial expression vector for GST-ATF-2 (residues 1–109) has been described (10). GST-c-Jun(1–82) and GST-JunB(1–138) were prepared using PCR and primers encompassing residues 1–82 of the amino-terminal region of mouse c-jun or primers encompassing residues 1–138 of mouse JunB. The PCR primers contained EcoRI sites such that the PCR product when cloned into the EcoRI site of pGEX2T (Pharmacia) in the proper orientation produced in-frame fusion protein. GST-JunB(1–97) was constructed from GST-JunB(1–138) by cleavage with BamHI and BclI and religation in the correct orientation into pGEX-2T. GST fusion proteins were purified on glutathione-agarose as described (21). Time-course solid-phase kinase assay of GST fusion proteins were performed as described (1). Approximately 100 μg of liver extracts was incubated with 2 μg of GST fusion protein bound to glutathione-agarose beads. The beads were washed extensively to remove unbound kinases and phosphorylation initiated by the addition of [γ-32P]ATP. Phosphorylated proteins were resolved on a 10% SDS/PAGE gel. JNK and p38 activity was measured using an immunecomplex kinase assay with recombinant GST-c-Jun(1–82), GST-ATF-2(1–109) and GST-JunB(1–138) as substrates as described (9).
Proteins were examined by Western blot analysis. From 100 to 200 μg of protein extract was electrophoresed on a 10% SDS/PAGE gel. The gel was soaked in protein transfer buffer (39 mM glycine/48 mM Tris base/0.037% SDS/20% methanol) for 20 min at room temperature and then transferred to nitrocellulose using a Trans-Blot semi-dry transfer cell (Bio-Rad). The blot was blocked with TBST (150 mM NaCl/50 mM Tris·HCl, pH 7.5/0.1% Triton X-100) with 5% nonfat dry milk. Blots were incubated with primary antibody in TBST with 5% milk overnight at 4°C, washed three times with TBST, and incubated 1 hr with goat anti-rabbit horseradish peroxidase (HRP)-conjugated antibody (Jackson ImmunoResearch). Signal was detected using ECL (DuPont/NEN).
Total RNA was isolated from CCl4-induced livers using Tripure reagent (Boehringer Mannheim) and hybridized to a [32P]-labeled antisense MKP-1 (22) RNA probe (452–589 bp), treated with T2 RNase, and analyzed on a 6% polyacrylamide/8 M urea gel as described (18). The 18S RNA protection assay is shown as a control for amount of RNA using a [32P]-labeled antisense probe for 18S RNA (Ambion, Austin, TX). MKP-1 mRNA levels were quantitated using a Molecular Dynamics PhosphorImager and normalized for 18S RNA. Liver tissue was prepared for cryostat sectioning by perfusion with 4% paraformaldehyde, followed by immersion overnight in 4% paraformaldehyde at 4°C. Tissues were cryoprotected by immersion in 30% sucrose overnight at 4°C, and then 7-μm sections were prepared on a cryostat and hybridized with 35S-labeled antisense RNA probe for mouse MKP-1 (22). Tissue sections were also hybridized with a negative control that was the sense strand of MKP-1. The sections were then washed and immersed briefly in Kodak NTB-2 emulsion. Slides were exposed, developed, and stained with standard hematoxylin/eosin stain. Sections were examined by bright- and dark-field microscopy (23).
RESULTS
CCl4-Induction of AP-1 Binding in the Liver.
The mechanism of activation of AP-1 target genes was examined in an EMSA using the c-Jun proximal AP-1 site as an oligonucleotide probe. We show that CCl4-induced oxidative stress causes the formation of several new AP-1 complexes (Fig. 1A, complexes A–C). The new complexes are first observed at 2 hr and become maximal by 4 hr. Induced complexes were also observed on other AP-1 target genes, such as the GST Ya AP-1 site (data not shown). The appearance of protein–DNA complexes (complexes A–C) precedes the appearance of c-Jun mRNA (10–24 hr; K.G.M. et al., unpublished data). To identify the components of the induced AP-1 binding complexes, we used antisera specific to several JUN/FOS family members and to ATF-2 in an EMSA (Fig. 1B). Since it has been shown that c-Jun can be involved in autoactivation of its own promoter through the proximal AP-1 site (24), we anticipated that c-Jun would be detected in one of the induced complexes (complexes A–C). Indeed, antisera to c-Jun appear to supershift complex C preferentially. Complex A appears to contain ATF-2 and JunD (immunodepletion), while complex B contains JunB (supershift) and c-fos (supershift). Finally, antisera to fra-1 do not appear to shift or deplete any of the induced or constitutive bands (data not shown). Although we cannot be certain we have identified all the components of the induced complexes, this analysis demonstrates that at least ATF-2, c-Jun, JunB, JunD, and c-fos are present in the activated complexes (complexes A–C) and are not present in the constitutive complexes (complexes D and E).
Figure 1.
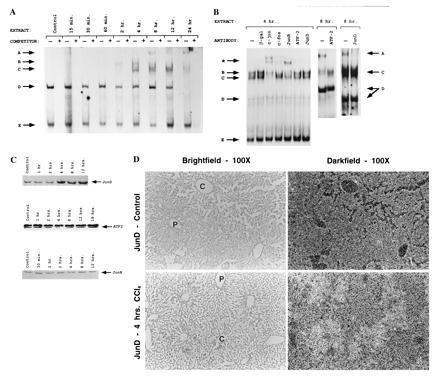
Metabolic oxidative stress activates new complex formation on AP-1 sites. (A) EMSA time course of AP-1 binding proteins after induction of oxidative stress with CCl4. Lanes − or + indicate absence or presence of 50-fold excess unlabeled oligonucleotide competitor. Arrows A–C indicate induced complexes, and arrows D and E indicate constitutive complexes. (B) Antibody supershift and immunodepletion of EMSA using 4-hr or 8-hr induced extract. Complexes A–D are as in A. The ∗ arrow indicates supershifted complexes. Supershifts were done with 4-hr extract because the location of induced band A present at 8 hr and later time points interferes with the observation of supershifted bands. (C) Western blot of Jun/ATF family members. About 200 μg of liver extracts from control or CCl4-treated mice were electrophoresed on 10% SDS/PAGE, blotted, and probed with antisera specific to ATF-2, JunB, and JunD (Santa Cruz Biotechnology). Arrows indicate specific immune complexes detected by enhanced chemiluminescence (DuPont). (D) Localization of JunD mRNA was determined using in situ hybridization. [35S]-labeled JunD antisense probe was hybridized to 7-μm sections of liver tissue from control and 4-hr CCl4-treated livers. The same field is shown using both bright-field and dark-field optics. P, portal vein; C, central vein. (×100.)
As an initial approach to determining the mechanism of activated AP-1 binding, we examined whether protein levels for several Jun family members increased in the liver after CCl4 treatment. As shown in Fig. 1C, Western blots of ATF-2 and JunB do not vary with time of treatment. Previously, we have shown that c-jun and c-fos do not change at early times (K.G.M. et al., unpublished data). Interestingly, JunD protein levels increased by 4 hr after CCl4 treatment. To determine if JunD activation was specific to pericentral hepatocytes undergoing oxidative stress, we examined the localization of JunD mRNA using in situ hybridization. As shown in Fig. 1D, JunD mRNA increased by 4 hr after CCl4 treatment and was localized to pericentral hepatocytes, as predicted. The role of JunD in transcriptional regulation has not been extensively characterized, although it has been implicated as an inhibitory AP-1 member and can partially block transformation by activated Ras (25, 26). Our data suggest that JunD may also play a role in the response of cells to stress.
Kinase Activation Precedes AP-1 Binding.
To examine the activation of the AP-1 binding proteins c-Jun and ATF-2, we first determined if there was increased kinase activity on the N-terminal activation domains of each protein using a solid-phase kinase assay. GST fusion proteins were incubated with control and induced extracts and the kinase activity of each extract was determined by the addition of [γ-32P]ATP. As shown in Fig. 2A, GST-c-Jun(1–82) is phosphorylated at a low level in control experiments, but by 1–2 hr after CCl4 treatment, phosphorylation of GST-c-Jun(1–82) is induced 5-fold. Similarly, GST-ATF-2(1–109) is phosphorylated at low levels in control experiments and increases 14-fold by 2 hr after CCl4 treatment (Fig. 2B). Interestingly, activation of c-Jun and ATF-2 kinases at 1–2 hr precedes the appearance of activated AP-1 binding activity. In addition, c-Jun and ATF-2 kinase activity remained elevated over an extended period (up to 8 hr, Fig. 2B). Finally, we considered whether JunB is phosphorylated by an activated kinase. JunB contains a Thr-Pro-Thr-Pro region (residues 102–105) that is similar to the phosphorylated region of ATF-2 (residues 69–72) and, therefore, may have similar regulation to ATF-2. We constructed an N-terminal GST-JunB fusion protein (residues 1–138) containing the putative phosphorylated region. As shown in Fig. 2C, GST-JunB(1–138) is phosphorylated at low levels in control experiments and at elevated levels 2 hr after CCl4 treatment.
Figure 2.
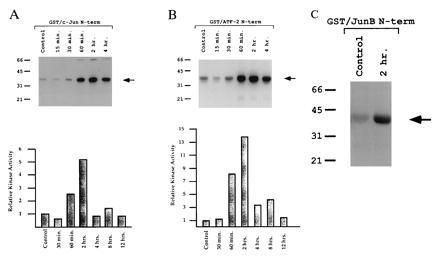
Metabolic oxidative stress activates JUN and ATF-2 kinases. (A) Time-course solid-phase kinase assay of GST-c-Jun(1–82) fusion protein. Liver extracts from CCl4-treated mice (times indicated) were incubated with GST-c-Jun(1–82) fusion protein bound to glutathione-agarose beads. The beads were washed extensively to remove unbound kinases and phosphorylation initiated by the addition of [γ-32P]ATP. Phosphorylated proteins were resolved on a 10% SDS/PAGE gel. Phosphorylated protein was quantitated using a Molecular Dynamics PhosphorImager. Relative kinase activity is plotted with phosphorylation by control extract standardized to 1 unit (y axis). The plotted data are the average of two experiments. (B) Time-course solid-phase kinase assay of GST-ATF-2(1–109) fusion protein performed as described in A. (C) Solid-phase kinase assay of GST-JunB(1–138) fusion protein performed using control and 2-hr extracts as described in A.
Independent Activation of JNK and Inactivation of p38.
The phosphorylation of ATF-2 and c-Jun (9, 10) suggests that JNK is activated by metabolic oxidative stress (Fig. 2 A and B). This was confirmed by examining the phosphorylation of c-Jun and ATF-2 by immunopurified JNK (Fig. 3A). JNK activity on GST-c-Jun(1–82) and GST-ATF-2(1–109) substrates was increased at 2 hr relative to control, similar to the observed solid-phase kinase activity. We also determined whether JunB is a substrate for JNK and whether the postulated phosphorylation region of JunB is required for JNK phosphorylation. As shown in Fig. 3B, GST-JunB(1–138) is phosphorylated by immunopurified JNK in the 2-hr activated extracts. We also prepared a construct, GST-JunB(1–97), that deletes the putative phosphorylated residues 102–105. Also shown in Fig. 3B, GST-JunB(1–97) is not a substrate for immunopurified JNK, showing that residues 102–105 are required for phosphorylation. Finally, JNK protein levels were measured by Western blot analysis and shown to be invariant in control and up to 18 hr after CCl4 treatment (Fig. 3C), indicating that JNK was specifically activated by metabolic oxidative stress. These data reconfirm the role of JNK in cellular stress responses and suggest that JunB is a new substrate for JNK and regulation by oxidative stress.
Figure 3.
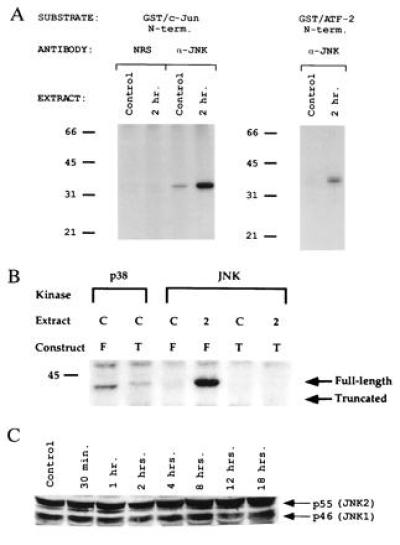
Regulation of JNK by metabolic oxidative stress. (A) JNK activation in CCl4-treated liver extracts. JNK activity was measured by an immunecomplex protein kinase assay using [γ-32P]ATP and GST-c-Jun(1–82) as a substrate. The phosphorylated GST-c-Jun(1–82) was detected after SDS/PAGE by autoradiography. NRS represents normal rabbit serum used as a negative control, and α-JNK represents anti-JNK antibody. (B) Determination of JunB phosphorylation site specificity. GST-JunB(1–138) or the truncated fusion protein GST-JunB(1–97) was used in immunecomplex kinase assays for either JNK or p38 MAP kinase. Extracts were used from control or 2-hr CCl4-treated livers. Notice that the band in the first lane is slightly smaller than the fusion protein as shown by Coomassie staining and does not represent phosphorylation of GST-JunB(1–138) fusion protein. T, truncated GST-JunB(1–97) fusion protein; F, full-length N-terminal GST-JunB(1–138) fusion protein. Arrows represent the positions of full-length (residues 1–138) or truncated (residues 1–97) fusion protein as determined by Coomassie staining. (C) Western blot of JNK family members. About 200 μg of liver extracts from control or CCl4-treated mice was electrophoresed on a 10% SDS/PAGE gel, blotted, and probed with antisera specific to JNK. Arrows indicate specific immune complexes to both JNK1 and JNK2 as detected by enhanced chemiluminescence (DuPont).
However, it was unclear from these experiments whether p38 played a role in the response of liver to metabolic oxidative stress. Therefore, we examined the phosphorylation of ATF-2 by immunopurified p38 (Fig. 4A). In control liver and at early time points after oxidative stress, no reproducible change in p38 protein kinase activity was detected. Unexpectedly, we observed a loss of p38 activity at 2 hr. The marked decrease in p38 kinase activity was sustained for at least 8 hr. p38 is activated by MKK3, MKK4, and MKK6 by dual phosphorylation on Thr-180 and Tyr-182 (9, 13, 14). To confirm the loss of p38 kinase activity, we examined the phosphorylation state of p38 in control and CCl4-treated liver. Phospho-p38 (p-p38) was examined by Western blot analysis using an anti-p-p38 antibody from New England Biolabs (Fig. 4B). p-p38 immunoreactivity was detected in extracts from control livers and up to 1 hr after CCl4 treatment. At 2 hr after CCl4 treatment, p-p38 immunoreactivity greatly decreased and was completely abolished from 4 to 8 hr, indicating that the mechanism of p38 inactivation was loss of phosphorylation. A Western blot of liver extract probed with anti-p38 antibodies showed similar levels of p38 protein in control and CCl4-treated livers, showing that the loss of activated p38 was not due to degradation of existing protein (Fig. 4B).
Figure 4.
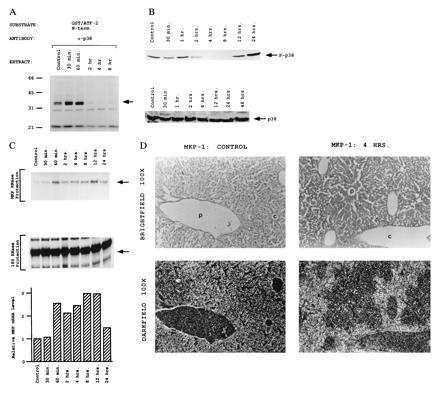
Constitutive activity of p38 MAP kinase in the liver is inactivated by metabolic oxidative stress. (A) p38 MAP kinase inactivation in CCl4-treated liver extracts. The activity of p38 MAP kinase was measured by an immunecomplex protein kinase assay using [γ-32P]ATP and GST-ATF-2(1–109) as a substrate. The phosphorylated GST-ATF-2(1–109) was detected after SDS/PAGE by autoradiography. (B) p38 MAP kinase phosphorylation during oxidative stress. About 200 μg of liver extracts from control or CCl4-treated mice was electrophoresed on a 10% SDS/PAGE gel, blotted, and probed with antisera specific to phospho-p38 (New England Biolabs) or with antisera specific to p38. Arrows indicate specific immune complexes to either phospho-p38 or p38 protein as detected by enhanced chemiluminescence (DuPont). (C) Activation of MKP-1 by metabolic oxidative stress. MKP-1 mRNA levels were analyzed by RNase protection assay. MKP-1 mRNA levels were quantitated using a Molecular Dynamics PhosphorImager and normalized for 18S RNA. The mean results of two experiments are shown. (D) Localization of MKP-1 mRNA was determined using in situ hybridization. [35S]-labeled MKP-1 antisense probe was hybridized to 7-μm sections of liver tissue from control and 4-hr CCl4-treated livers. The same field is shown using both bright-field and dark-field optics. P, portal vein; C, central vein. (×100.)
Mechanism of p38 Dephosphorylation/Inactivation.
Two possible mechanisms could account for the observed inactivation of p38. (i) Since p38 appears to be constitutively active in the liver, one of the known p38 upstream regulators, such as MKK3 or MKK6, might be repressed after CCl4 treatment. However, since both p38 and JNK are substrates for MKK4 (14, 15), inactivation of the upstream signaling pathway may not account for the differential regulation of the JNK and p38 MAP kinases in oxidatively stressed liver. (ii) There could be activation of a phosphatase that inactivates p38. The dual specificity phosphatase MKP-1 has recently been shown to be a potent regulator of both p38 and ERK1,2 (9, 19, 27, 28). In contrast, MKP-1 weakly inactivates JNK (27, 28, 30). Furthermore, MKP-1 is strongly induced by JNK activation caused by constitutively active MEKK1 and by environmental stress (29–31). We therefore determined whether MKP-1 is induced by metabolic oxidative stress. Induction of MKP-1 mRNA was detected within 1 hr in an RNase protection assay (Fig. 4C). Because MKP-1 can affect both ERK and p38, we examined the localization of MKP-1 induction in liver using in situ hybridization (Fig. 4D). MKP-1 mRNA was specifically induced in pericentral hepatocytes that are undergoing oxidative stress. Furthermore, the restricted expression of MKP-1 in pericentral hepatocytes (no more than 50% of the total hepatocytes) suggests that the 2.5-fold induction of MKP-1 mRNA in total liver (Fig. 4C) represents an actual increase of at least 5-fold in pericentral hepatocytes. These data demonstrate that, in addition to being activated by mitogens and extracellular stress, MKP-1 is also induced by metabolic oxidative stress and regulates p38 activity.
DISCUSSION
Cellular signaling by proinflammatory cytokines and extracellular stress activates the JNK and p38 MAP kinase family members. A major target of the activated kinases is the AP-1 transcription factor (32), which is composed of homo- and heterodimeric complexes of JUN and FOS proteins. Members of the JUN family can also form heteromeric complexes with ATF proteins (33). In the case of c-Jun and ATF-2, regulation is conferred by direct phosphorylation within the activation domain of each transcription factor. Interestingly, there are substrate specificity differences between JNK and p38: JNK will phosphorylate c-Jun and ATF-2, while p38 phosphorylates only ATF-2 (9, 10). We have extended this substrate specificity to show that JunB is phosphorylated by JNK but not by p38 (Fig. 3B).
In addition to the different substrate specificities of JNK and p38, the upstream MKK-activating proteins show specificity with respect to regulation of JNK and p38; MKK4 activates both JNK and p38, while MKK3 and MKK6 activate only p38. These differences suggest that there could be both convergence and divergence in the regulation of the JNK and p38 pathways. To our knowledge, our data illustrate this for the first time by demonstrating that p38 is constitutively active in the liver and is down regulated by oxidative stress (Fig. 4 A and B). The kinetics of the independent regulation of JNK and p38 show that maximal JNK activation occurs at the same time as p38 repression occurs (2 hr, Figs. 2, 3, 4). Therefore, though MKK4/SEK is likely activated (leading to the activation of JNK), the concomitant activation of MKP-1 appears to override any potential MKK4/SEK signal to p38 at 2 hr. Furthermore, since p38 is apparently constitutively active, an additional positive signal deriving from the activation of MKK4/SEK may be difficult to detect in the presence of the high background activity. The importance of MKP-1 as a potential regulator of p38 in stressed liver is supported by reports indicating that MKP-1 may be a more potent regulator of p38 MAP kinase than of the ERK group of MAP kinases (9, 30, 34). The identification of constitutively active p38 MAP kinase in the liver and the independent regulation of p38 and JNK represent a new aspect of these stress-induced signaling pathways (Fig. 5). The consequences of p38 MAP kinase suppression on gene expression will be particularly interesting as more substrates for p38 MAP kinase are identified.
Figure 5.
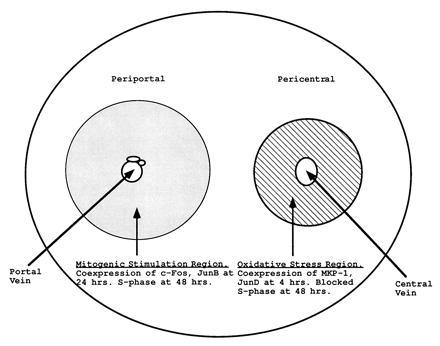
Model for the independent regulation of JNK and p38 kinase pathways in the liver. p38 MAP kinase is constitutively active in the liver. Oxidative stress dependent on the pericentral localization of P450 enzymes leads to the activation of JNK and the subsequent phosphorylation of transcription factor substrates such as c-Jun and ATF-2. Furthermore, transcription factors such as JunD are also induced in the pericentral region. Finally, metabolic oxidative stress induces MKP-1 in the pericentral region, perhaps via activation of the JNK signal transduction pathway (30). The induction of MAP kinase phosphatase can suppress the constitutive activity of p38 MAP kinase (9, 22).
The activation of MKP-1 in pericentral hepatocytes is likely to have other consequences for cellular regulation. MKP-1 inhibits activated Ras induction of DNA synthesis in transfected cells (27). We (K.G.M. et al., unpublished data) and others (35, 36) have observed that pericentral hepatocytes do not enter S phase after oxidative stress to the liver, although periportal hepatocytes can enter the cell cycle. We suggest that induction of MKP-1 in stressed hepatocytes may be important in preventing DNA replication in cells that may be damaged. This possibility is intriguingly supported by the coexpression of JunD and MKP-1 in pericentral hepatocytes. It has previously been shown that JunD is an inhibitory member of the AP-1 family (25). Furthermore, JunD was shown to accumulate in quiescent cells and to be able to partially block transformation by activated Ras (26). Finally, stable overexpression of JunD results in an increased number of cells in G1 phase, suggesting that JunD can slow entry into the cell cycle (26). Thus at least two proteins linked to suppression of cell cycle (MKP-1 and JunD) are activated in pericentral hepatocytes in response to oxidative stress, possibly leading to the observed decrease in the replicative capacity of these cells.
The induction of proteins that can suppress the cell cycle in hepatocytes undergoing oxidative stress is interesting in light of evidence linking JNK with ceramide-mediated apoptosis (37, 38) and with induction of apoptosis in neuronal cells (39). Our observation of prolonged activation of JNK in stressed hepatocytes may be linked to the subsequent cell death observed in the pericentral region. This is consistent with the neuronal cell apoptosis paradigm because although nerve growth factor (NGF) withdrawal activates both p38 and JNK, constitutive activation of p38 only or JNK only is sufficient to induce apoptosis. Therefore, the ability of cells to independently regulate JNK/p38 MAP kinase signaling may provide additional checkpoints in the control of cellular apoptosis and other cellular processes.
Acknowledgments
We thank Drs. A. S. Yee, B. Cochran, and L. Feig for helpful discussions and review of this manuscript. We also thank Dr. N. J. Tonks for providing the MKP-1 cDNA and Dr. R. Bravo for providing the mouse c-Jun and JunB cDNAs. R.J.D. is an investigator of the Howard Hughes Medical Institute. This work was supported by National Institutes of Health Grants RO1 DK 50442 (K.E.P.), RO1 CA 65861 (R.J.D.), and RO1 CA 58396 (R.J.D.). This project was also funded at least in part with federal funds from the U.S. Department of Agriculture, Agricultural Research Service under Contract 53-3K06-01 (K.E.P.).
Footnotes
Abbreviations: MAP kinase, mitogen-activated protein kinase; MKK, MAP kinase kinase; GST, glutathione S-transferase; EMSA, electrophoretic mobility-shift assay.
References
- 1.Hibi M, Lin A, Smeal T, Minden A, Karin M. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 2.Dérijard B, Hibi M, Wu I-H, Barrett T, Su B, Deng T, Karin M, Davis R J. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 3.Sluss H K, Barrett T, Dérijard B, Davis R J. Mol Cell Biol. 1994;14:8376–8384. doi: 10.1128/mcb.14.12.8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kyriakis J M, Banerjee P, Nikolakaki E, Dai T, Rubie E A, Ahmad M F, Avruch J, Woodget J R. Nature (London) 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 5.Galcheva-Gargova Z, Dérijard B, Wu I-H, Davis R J. Science. 1994;265:806–808. doi: 10.1126/science.8047888. [DOI] [PubMed] [Google Scholar]
- 6.Minden A, Lin A, Smeal T, Dérijard B, Cobb M, Davis R, Karin M. Mol Cell Biol. 1994;14:6683–6688. doi: 10.1128/mcb.14.10.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kallunki T, Su B, Tsigelny I, Sluss H K, Dérijard B, Moore G, Davis R J, Karin M. Genes Dev. 1994;8:2996–3007. doi: 10.1101/gad.8.24.2996. [DOI] [PubMed] [Google Scholar]
- 8.Han J, Lee J-D, Bibbs L, Ulevitch R J. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 9.Raingeaud J, Gupta S, Rogers J S, Dickens M, Han J, Ulevitch R J, Davis R J. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 10.Gupta S, Campbell D, Dérijard B, Davis R J. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 11.Livingstone C, Patel G, Jones N. EMBO J. 1995;14:1785–1797. doi: 10.1002/j.1460-2075.1995.tb07167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whitmarsh A J, Shore P, Sharrocks A D, Davis R J. Science. 1995;269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]
- 13.Raingeaud J, Whitmarsh A J, Barrett T, Dérijard B, Davis R J. Mol Cell Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dérijard B, Raingeaud J, Barrett T, Wu I-H, Han J, Ulevitch J, Davis R J. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 15.Lin A, Minden A, Martinetto H, Claret F-X, Lange-Carter C, Mercurio F, Johnson G L, Karin M. Science. 1995;268:286–290. doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- 16.Sanchez I, Hughes R T, Mayer B J, Yee K, Woodget J R, Avruch J, Kyriakis J M, Zon L I. Nature (London) 1994;372:794–798. doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- 17.Wolf C R, Moll E, Oesch F, Buchmann A, Kuhlmann W D, Kunz H W. Carcinogenesis. 1984;5:993–1001. doi: 10.1093/carcin/5.8.993. [DOI] [PubMed] [Google Scholar]
- 18.Pimental R A, Liang B, Yee G K, Wilhelmsson A, Poellinger L, Paulson K E. Mol Cell Biol. 1993;13:4365–4373. doi: 10.1128/mcb.13.7.4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ward Y, Gupta S, Jensen P, Wartmann M, Davis R J, Kelly K. Nature (London) 1994;367:651–654. doi: 10.1038/367651a0. [DOI] [PubMed] [Google Scholar]
- 20.Cuthill S, Wilhelmsson A, Poellinger L. Mol Cell Biol. 1991;11:401–411. doi: 10.1128/mcb.11.1.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith S B, Johnson K S. Gene. 1988;67:31–39. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 22.Charles C H, Sun H, Lau L F, Tonks N K. Proc Natl Acad Sci USA. 1993;90:5292–5296. doi: 10.1073/pnas.90.11.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuo C F, Paulson K E, Darnell J E., Jr Mol Cell Biol. 1988;8:4966–4971. doi: 10.1128/mcb.8.11.4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Angel P, Hattori K, Smeal T, Karin M. Cell. 1988;55:875–885. doi: 10.1016/0092-8674(88)90143-2. [DOI] [PubMed] [Google Scholar]
- 25.Metiveir C, Piu F, Pfarr C, Yaniv M, Loiseau L, Castellazzi M. Oncogene. 1993;8:3211–3215. [PubMed] [Google Scholar]
- 26.Pfarr C, Mechta F, Spyrou G, Lallenmand D, Carillo S, Yaniv M. Cell. 1994;76:747–760. doi: 10.1016/0092-8674(94)90513-4. [DOI] [PubMed] [Google Scholar]
- 27.Sun H, Tonks N K, Bar-Sagi D. Science. 1994;266:285–288. doi: 10.1126/science.7939666. [DOI] [PubMed] [Google Scholar]
- 28.Sun H, Charles C H, Lau L F, Tonks N K. Cell. 1993;75:487–493. doi: 10.1016/0092-8674(93)90383-2. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Gorospe M, Yang C, Holbrook N J. J Biol Chem. 1995;270:8377–8380. doi: 10.1074/jbc.270.15.8377. [DOI] [PubMed] [Google Scholar]
- 30.Bokemeyer D, Sorokin A, Yan M, Ahn N G, Templeton D J, Dunn M J. J Biol Chem. 1996;271:639–642. doi: 10.1074/jbc.271.2.639. [DOI] [PubMed] [Google Scholar]
- 31.Keyse S M, Emslie E M. Nature (London) 1992;359:644–647. doi: 10.1038/359644a0. [DOI] [PubMed] [Google Scholar]
- 32.Karin M. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 33.Kerppola T K, Curran T. Curr Opin Struct Biol. 1991;1:71–79. [Google Scholar]
- 34.Alessi D R, Gomez N, Moorhead G, Lewis T, Keyse S M, Cohen P. Curr Biol. 1995;5:283–295. doi: 10.1016/s0960-9822(95)00059-5. [DOI] [PubMed] [Google Scholar]
- 35.Gebhardt, R. (1988) Scand. J. Gastrenterol. 23, Suppl. 151, 8–18.
- 36.Gebhardt R, Burger H-J, Heini H, Schreiber K-L, Mecke D. Hepatology. 1988;8:822–830. doi: 10.1002/hep.1840080421. [DOI] [PubMed] [Google Scholar]
- 37.Verheij M, Bose R, Lin X H, Yao B, Jarvis W D, Grant S, Birrer M J, Szabo E, Zon L I, Kyriakis J M, Haimovitz-Friedman A, Fuks Z, Kolesnick R N. Nature (London) 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 38.Cuvillier O, Pirianov G, Kleuser B, Vanek P G, Coso O A, Gutkind J S, Spiegel S. Nature (London) 1996;381:800–803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- 39.Xia Z, Dickens M, Raingeaud J, Davis R J, Greenberg M E. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]