

Abstract
A novel RNase activity was identified in a yeast RNA polymerase I (pol I) in vitro transcription system. Transcript cleavage occurred at the 3′ end and was dependent on the presence of ternary pol I/DNA/RNA complexes and an additional protein factor not identical to transcription factor IIS (TFIIS). Transcript cleavage was observed both on arrested complexes at the linearized ends of the transcribed DNA and on intrinsic blocks of the DNA template. Shortened transcripts that remained associated within the ternary complexes were capable of resuming RNA chain elongation. Possible functions of the nuclease for transcript elongation or termination are discussed.
Keywords: in vitro transcription, ternary complex, RNA cleavage, transcriptional arrest, Saccharomyces cerevisiae
RNA polymerase-associated transcript cleavage has recently emerged as a common feature of several prokaryotic and eukaryotic transcriptionally active enzyme complexes. Hydrolytic transcript cleavage was first demonstrated in Escherichia coli RNA polymerase ternary complexes (1): two E. coli transcription elongation factors, GreA and GreB, mediate RNA cleavage of nascent transcript followed by the loss of the 3′ proximal fragment and resumption of elongation from the new 3′ terminus (2–4). It is proposed that certain DNA sequences through which the elongating RNA polymerase has to pass lead to paused transcription complexes that can spontaneously convert into nonextensible, dead-end conformations and result in arrest of RNA chain elongation (5). Relief of dead ends and restart of elongation appear to be mediated by the 3′ proximal cleavage of the transcripts by GreA or GreB to restore the RNA 3′ terminus to the catalytic center of the RNA polymerase (4, 6).
In eukaryotes, hydrolytic cleavage in transcriptionally active enzyme complexes has been shown to be associated with RNA polymerases (pol) I, II, and III. The pol II elongation complex utilizes a mechanism similar to the prokaryotic RNA polymerase to extend blocked transcripts with the help of the elongation factor TFIIS (transcription factor IIS) (7–9). In the presence of TFIIS the ternary complex cleaves up to 14 nt from the RNA in a 3′–5′ manner, releasing predominantly mono- and dinucleotides (10–13). This cleavage seems to be a prerequisite for TFIIS-mediated transcription through blocks of RNA chain elongation (11, 12, 14–16).
Although cleavage is highly dependent on the presence of TFIIS, the particular nucleolytic activity probably does not reside in this accessory factor. It seems likely that pol II possesses intrinsic hydrolytic activity, which is activated by the elongation factor (9, 13, 15, 17, 18). Recently, a 3′–5′ exonuclease activity that is associated with yeast pol III ternary complexes was identified and shown not to depend on the presence of auxiliary proteins such as TFIIS (19).
An exonuclease activity associated with pol I was described in a mammalian pol I transcription termination complex. This complex utilizes a DNA binding protein, TTFI (transcription termination factor I), to terminate pol I transcription. After termination, the 3′ end of the pre-rRNA is shortened by 10 nucleotides in vivo and in vitro. Since TTFI alone has no hydrolytic activity, the nuclease presumably resides in pol I (20, 21). Two subunits of yeast pol I, A49 and A40, have been shown independently to possess RNaseH activity (22, 23). So far, all described pol I-specific hydrolytic nuclease activities do not appear to require a ternary transcription complex, because their nucleic acid subtrates are also cleaved posttranscriptionally.
To screen for rRNA modulating activities, an in vitro transcription system was used that generates accurately initiated pol I-dependent transcripts. An exonuclease activity that removes up to 14 nt from the 3′ end of arrested transcripts could be identified. The RNA cleavage activity is specific for pol I, functions in a ternary complex, and depends on the presence of a dissociable factor distinguishable from TFIIS.
MATERIALS AND METHODS
Plasmids and Yeast Strains.
Saccharomyces cerevisiae strain BJ926 was used for pol I-dependent transcription and for the preparation of extracts containing RNA cleavage activity. Control strain for disrupted mitochondrial RNA polymerase was yJJ189 (24). Templates for transcription were derived from pSES5 (25) and were linearized by EcoRV, BamHI, or SalI. If not indicated elsewhere, the pSES5/EcoRV fragment was used for in vitro transcription reactions. To construct plasmid pSKpI, the pSES5 HindIII/EcoRV fragment was inserted into puc 18. The DNA containing the pol I promoter was cut with PvuII and HindIII, and the resulting fragment of ≈550 bp was ligated into a HindIII/SmaI-cleaved pBluescript SK vector. For generation of in vitro transcripts the vector was linearized by BamHI, SacI, or SacII.
To attach magnetic beads, pSES5 was linearized by HindIII, which cuts 459 bp upstream of the start site of RNA synthesis. After the HindIII site was filled in with biotinylated dATP and dCTP using the Klenow fragment of DNA polymerase, the plasmid was cut with EcoRV and the resulting 800-bp fragment was isolated. Magnetic beads [25 μl Dyna beads (Dynal, Great Neck, NY), 6 × 108–7 × 108 beads/ml] were washed three times with buffer TEBCl (50 mM Tris·HCl, pH 7.5/1 mM EDTA/1 M NaCl/0.1 mg/ml bovine serum albumin), and 3 μg DNA was added to a total volume of 100 μl TEBCl. After washing three times with TEBCl, the beads were resuspended in 100 μl water.
To construct the 3′-extended template pItailKS, the two oligonucleotides 5′-TAGGAGAGGTGTGAGGAGAGGTTGATGAAAGTGTATAAGCTTT-3′ (template strand) and CTAGAAAGCTTATACACTTTCATCAACCTCTCCTCACACCTCTCCTACCAAATTCCACCACATTTCCAATAT-biotine 3′ were hybridized and ligated to the XbaI/PvuII 145-bp fragment of pBluescript KS and attached to magnetic beads as previously described. Transcription started 14 nt upstream of the junction to produce a 53-nt-long RNA fragment when CTP was omitted from the transcription buffer. General molecular biological methods were as described (26).
In Vitro Transcription.
Run-off transcription was performed as described (30), with the exception that TRX (20 mM Hepes/KOH, pH 7.8/10 mM MgCl2/5 mM EGTA/0.1 mM EDTA/2.5 mM DTT/200 mM potassium acetate/0.2 mM ATP/0.2 mM CTP/0.2 mM UTP/0.01 mM GTP/3 μCi [32P]GTP; 1 Ci = 37 GBq) was used as transcription buffer. If radiolabeled transcripts were to be isolated, the completed transcription reaction was treated with 1.5 units RNase-free DNase for 10 min at 30°C. After proteinase K treatment and ethanol precipitation, the transcripts were dissolved in 3 μl H2O, heated to 70°C for 5 min, and added to transcription reactions.
Preparation of Pol I-Containing Initiation Complex.
For preparation of transcription extracts the basic protocol to reveal pol II-dependent transcription (28) was followed with some modifications. After 100,000 × g centrifugation, the supernatant was dialized against buffer A [20% glycerol/20 mM Hepes, pH 7.8/10 mM MgCl2/0.2 mM EDTA/1 mM DTT/1 mM phenylmethylsulfonyl fluoride (PMSF)/2 mM benzamidine] and applied to a DEAE Sepharose column (13 × 5.2 cm). The column was washed with 1 liter A90 (buffer A supplemented with 90 mM KCl), and protein was eluted with A350 (350 mM KCl) to reveal the described pol I-containing initiation complex (DEAE 0.35 fraction) (29). For further purification, fraction DEAE 0.35 was dialized against buffer B (20% glycerol/20 mM Hepes, pH 7.8/2 mM MgCl2/0.02 mM EDTA/1 mM DTT/1 mM PMSF/2 mM benzamidine) and subsequently centrifuged at 20,000 rpm in a Kontron TFT 50.38 rotor for 30 min. The pellet that contained all pol I-dependent transcriptional activity was resuspended in buffer B600 (buffer B containing 600 mM potassium acetate) at a concentration of 2.5 mg/ml to give fraction PA600. PA600 (0.5 μl) was used in a regular transcription assay for pol I-specific transcription.
Preparation of Mitochondrial Polymerase.
To isolate initiation-competent mitochondrial polymerase the same whole cell extract was used as described for the generation of pol I-dependent transcription. It was shown that the flowthrough of the DEAE column (A90) contains both mitochondrial polymerase RPO41 and initiation factor MTF1 (ref. 29; unpublished data) and therefore is able to initiate transcription from the mitochondrial promoter consensus sequence 85 bp upstream of the pol I transcription start site (30). The same fraction prepared from a strain lacking the catalytic subunit of mitochondrial polymerase yJJ189 was not able to support transcription from this promoter-like sequence (ref. 30; data not shown). The mitochondrial transcription complex was further purified. It was applied to a Bio-Rex 70 column (9.5 cm × 5.2 cm) (Bio-Rad) washed with buffer B300 and eluted with B600. After dialysis against buffer B the active fractions were loaded on a Resource Q column (6-ml bead volume) (Pharmacia) and eluted by a linear gradient (120 ml) from buffer B100 to B600 (flow rate, 2 ml/min). The initiation-competent transcription complex eluted with 130 mM potassium acetate from the column. The peak fraction (2 μl) was used for in vitro transcription assays.
Purification of Cleavage Factor.
Preparation of whole cell extract and chromatography on Bio-Rex 70 and DEAE Sepharose was as described (31) with the exception of using DEAE Sepharose instead of DE52. The cleavage activity was detected in the flow-through of the DEAE Sepharose column. Active fractions were loaded on a Resource S column (6 ml). The column was washed with 30 ml of B100, and cleavage activity was eluted with a linear gradient (120 ml) from B100 to B600 (flow rate 2 ml/min). Activity eluted around 400 mM potassium acetate.
Primer Extension Analysis.
Run-off transcripts were synthesized in vitro in the presence of 0.2 mM GTP instead of [32P]GTP with or without 3 μl fraction 41. After proteinase K treatment and precipitation the pellet was dried and 30,000 cpm 32P-labeled oligonucleotide (on 5-115) 5′-TTTAACTGTGATAAACTACC-3′, located 115 bp downstream of the pol I transcription start site in reverse direction, was added. Reverse transcription was performed as described (32). The same oligonucleotide served as primer for a DNA sequencing reaction using pSES5 as the template. A sequencing gel was used to separate the resulting DNA fragments.
S1-Nuclease Mapping.
In the presence or absence of 3 μl fraction 41 radiolabeled in vitro, transcripts were hybridized to 50 pmol oligonucleotide S-13p (186–249 bp downstream of the pol I transcription start site in reverse direction) and subjected to S1-nuclease analysis as described (32). Protected fragments were separated on a 13% polyacrylamide gel containing 7 M urea.
Immobilized Transcription Assay.
Promoter-specific run-off transcriptions were performed with 30 ng template bound to magnetic beads. After 20 min at 25°C the supernatant was separated from the magnetic beads, the beads were washed twice with ice cold buffer TRX and resuspended in 25 μl TRX, and 3 μl of cleavage factor was added to both the bead fraction and the supernatant and incubated for an additional 30 min at 25°C. Transcription on immobilized 3′ extended template (pItailKS) (40 ng/reaction) was initiated in buffer TRX in the presence of 3 μCi [32P]GTP with 4 μl PA600. After 25 min at 25°C the magnetic beads were washed twice with 0.5 M potassium acetate in TRX without NTP and resuspended in 4 mM Tris·HCl, pH 8/4 mM KCl/0.04 mM EDTA/3.2% glycerol/0.16 μg/ml acetylated BSA/4 mM 2-mercaptoethanol/7 mM MgCl2. Fraction 41 (3 μl) was added (final volume, 25 μl) and incubated for 20 min at 30°C. To reextend the transcripts the magnetic beads were washed again with TRX without NTP and resuspended in 25 μl TRX with NTP for 20 min at 25°C. The resulting transcripts were analyzed as previously described.
Immunoprecipitation.
Fraction 41 (50 μl) was immunodepleted with 2 μl of immunserum and 2 × 20 μl of protein A Sepharose as described (33), using a slightly modified procedure.
RESULTS
Pol I-Specific Transcripts Are Shortened by a Protein Factor.
To establish a pol I in vitro transcription assay that allows accurate initiation from the yeast ribosomal promoter, whole cell extracts of S. cerevisiae were fractionated and the fractions were analyzed in run-off transcription assays. The template used in the run-off transcription assays, linearized pSES5, contained a pol I enhancer and the initiation site of the 35S rRNA and has been shown to support a high level of transcription in vivo (25) and in vitro (34). RNA synthesis initiated accurately on the ribosomal promoter as verified by primer extension analyses (see Fig. 1A).
Figure 1.
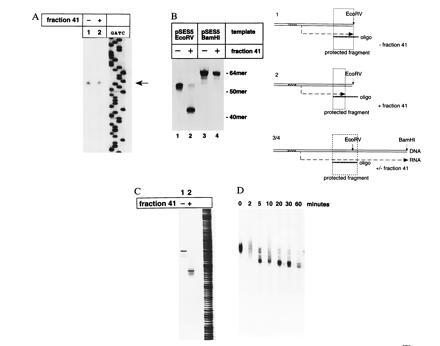
Transcripts are cleaved from the 3′ end. (A) Primer extension. Pol I-dependent transcripts were generated in the absence (lane 1) or presence (lane 2) of fraction 41 (Resource S column) and reverse-transcribed using a 32P-labeled oligonucleotide (on 5-115) as the primer. A DNA sequencing reaction with the same oligonucleotide as the primer and pSES5 as the template was performed independently. The resulting DNA fragments were separated on a 7% polyacrylamide sequencing gel. The correct start site of pol I-dependent transcription is indicated by an arrow. (The first 8 nt are 5′-AUGCGAAA.) (B) S-1 nuclease mapping of the 3′ end. Transcription from pSES5 linearized with the enzymes indicated was performed in the presence of [32P]GTP. Isolated radiolabeled RNA was hybridized to oligonucleotide S-13p and subjected to S-1 nuclease treatment. Protected fragments were visualized by autoradiography of 13% polyacrylamide/7 M urea gels. As schematically detailed, RNA transcribed from the BamHI-linearized pSES5 and hybridized to the oligonucleotide should lead to a completely protected fragment 64 nt long (lanes 3 and 4, respectively, and 3/4 in the scheme). In contrast, a shorter fragment should result from the hybridization of the same oligonucleotide with RNA transcribed from the EcoRV-linearized template (lane 1 and 1 in the scheme). If RNA cleavage takes place at the 3′ end of the transcript, the addition of fraction should lead to an even shorter protected fragment (lane 2 and 2 in the respective representation). Positions of marker oligonucleotides are indicated at the right. (C) Analysis of run-off transcripts on a high-resolution gel. Accurate initiated transcripts were synthesized in vitro in the absence (lane 1) or presence (lane 2) of fraction 41 and separated on a 7% denaturing polyacrylamide sequencing gel. A purin/pyrimidine ladder is shown on the right. (D) Time course of transcript cleavage. After the generation of full-length transcripts by the pol I complex for 20 min, fraction 41 was added. Equal amounts of the reaction were removed and stopped at time points indicated.
Run-off transcription performed with a crude pol I preparation (DEAE 0.35 fraction; ref. 29) resulted in a heterogenous population of transcripts, whereas transcripts generated by the further purified PA600 fraction were more homogenous in length (data not shown). PA600 contained the pol I initiation complex, was about 300-times enriched, and was not contaminated with other RNA polymerases (unpublished data). To analyze whether the observed reduction in RNA heterogeneity was due to the loss of an enzymatic activity during the purification of the pol I initiation complex, a yeast whole cell extract was fractionated and screened for activities that influence the transcript length produced by the PA600 fraction. This led to the identification of an activity that shortens the run-off transcripts. The activity was further purified by three chromatography steps (see Materials and Methods). The final preparation obtained by chromatography on Resource S did not contain detectable traces of any RNA polymerase, as tested by nonspecific RNA polymerase assays and by Western blotting (data not shown). For all further experiments, fraction 41 of the Resource S chromatography, which represented an approximate 800-fold enriched protein fraction, was used. Since RNA shortening was due to the addition of a partially purified protein fraction, it seemed unlikely that the processing activity resulted from pyrophosphorolysis. Pyrophosphorolysis, the reversal of elongation, is carried out in vitro by template-engaged enzymes in the presence of millimolar levels of pyrophosphate and generates truncated transcripts in ternary complexes with any kind of polymerase (8). To exclude the possibility that fraction 41 by itself generates excessive pyrophosphate under transcriptional conditions, fraction 41 was incubated with the template and transcription buffer for 30 min and subsequently treated with proteinase K (data not shown). After inhibition of the protease with PMSF, the proteinase K-treated fraction did not affect the transcript length further (data not shown). These results showed that the RNA shortening activity is associated with a protein factor.
A Total of 10-14 nt Are Removed from the 3′ End of a Completed Transcript.
Primer extension analysis and S-1 nuclease mapping were performed to determine which end of the RNA was affected by the shortening reaction. Fraction 41 did not influence the correct start site of ribosomal RNA synthesis (Fig. 1A); however, it did shorten the 3′ end of the synthesized transcript (Fig. 1B). Using high resolution sequencing gels, the precise distribution of the run-off transcripts was determined. In the presence of fraction 41 the synthesized RNA is 10-14 nt shorter (Fig. 1 C and D) than in reactions where fraction 41 was omitted. To distinguish whether the transcript truncation was due to a premature stop of the elongating pol I or to an enzymatic activity that hydrolyzes part of the completed transcript, the following experiment was performed. Under the conditions described, generation of accurate initiated run-off transcripts by fraction PA600 was completed after 20 min. Extended incubation times did not yield more radiolabeled transcripts. When fraction 41 was added after transcription was completed, removal of the terminal nucleotides occured within 5 min (Fig. 1D). No further remarkable cleavage product could be observed even after prolonged incubation times up to 1 h.
RNA Cleavage Is Not Sequence Specific.
A similar cleavage pattern was obtained when different linearized plasmids containing the ribosomal promotor were employed as templates for specific initiated RNA synthesis. Transcription assays in the presence of fraction 41 with templates cut by six different restriction enzymes revealed removal of the terminal 8–14 nt. However, the cleavage efficiency apparently depended on some feature intrinsic to the respective linearized end (Fig. 2; data not shown). By far the most efficient cleavage was obtained with the EcoRV fragment of pSES5.
Figure 2.
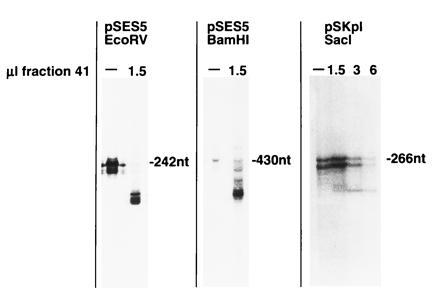
RNA cleavage is not sequence-specific. Transcription assays with different run-off templates. Plasmids pSES5 and pSKpI were linearized with different restriction enzymes as indicated. The resulting DNA served as templates for promoter-specific pol I-dependent transcription in the absence or presence of fraction 41. The resulting transcripts were separated on a 7% polyacrylamide sequencing gel. Note that EcoRV, BamHI, and SacI produce blunted, 5′, and 3′ overhung ends on the templates.
RNA Cleavage Is Restricted to Nascent Transcripts That Are Part of the Ternary Complex.
The efficiency of pol I-specific transcript cleavage depended not only on the nature of the DNA end but also on the quality of the transcribing pol I complex (compare Fig. 3A Upper, lane 2 with the time course experiment shown in Fig. 1D). For instance, when the time course experiment described in Fig. 1D was repeated with an independently derived PA600 protein fraction, a substantial amount (up to 50%) of the completed run-off transcripts remained uncleaved (data not shown). Neither a large excess of fraction 41 nor extended incubation times resulted in a quantitative cleavage of the full-length transcripts. If the cleavage mechanism requires that the transcript is still associated with DNA and protein factors, uncleaved RNA that has been released would not be a suitable substrate for the cleavage factor and could account for unprocessed RNA seen in Fig. 3A Upper, lane 2. To distinguish between bound and released transcripts, templates for the transcription reaction were coupled to magnetic beads. After 20-min synthesis of promoter-specific run-off transcripts, the supernatant was separated from the magnetic beads. In the experiment shown in Fig. 3A, ≈57% of the generated transcripts were released into the supernatant when transcription was performed. Only transcripts that were still bound to the template were cleaved in the presence of fraction 41, whereas free RNA appeared to be completely unaffected (Fig. 3A Upper, lanes 3 and 4). The requirement of a ternary RNA/DNA/protein complex for the cleavage activity was shown in the following experiment. 32P-labeled run-off transcripts, derived from transcription with the pSES5/BamHI fragment as a template, were isolated and added to transcription reactions that contained the pSES5/EcoRV fragment as a template. Only the nascent transcript was accessible to cleavage by fraction 41 (Fig. 3A Lower). To rule out the involvement of an RNase H activity, radiolabeled transcripts were isolated, hybridized to their corresponding template, and incubated with PA600 and fraction 41 under transcription conditions in the absence of labeled nucleotides. None of the labeled transcripts could be cleaved, whereas RNase H was able to hydrolyze the labeled RNA in the DNA/RNA hybrid (data not shown).
Figure 3.
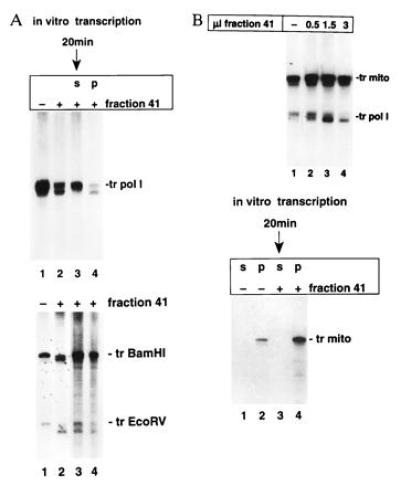
A ternary complex is essential to enable RNA cleavage in pol I-dependent transcription assays. (A Upper) Transcript cleavage reaction is restricted to ternary complexes. Run-off transcription was performed with immobilized template for 20 min. After separating the supernatant from the pellet, fraction 41 was added to both fractions (lanes 3 and 4). In control reactions fraction 41 was added (lane 2) or omitted (lane 1) to transcription assays with completed transcripts generated from DNA template in solution. The position of uncleaved transcripts is indicated (tr pol I). (A Lower) 32P-labeled run-off transcripts generated from the pSES5/BamHI fragment were isolated after DNase and proteinase K treatment. Aliquots were added to transcription reactions containing pSES5/EcoRV as the template in the presence of 1.5 μl (lane 3) and 3 μl (lane 4) of fraction 41. Control reactions show transcripts derived from the pSES5/BamHI and pSES5/EcoRV template when processed in the absence (lane 1) or presence (lane 2) of fraction 41. Positions of the different uncleaved transcripts are indicated (tr BamHI and tr EcoRV). (B Upper) Cleavage factor distinguishes between two different cotranscriptionally synthesized RNA. Increasing amounts of fraction 41 (lanes 2–4) were added to specific initiated run-off transcription assays in which mitochondrial RNA polymerase and pol I simultanously transcribed the same template. Positions of the corresponding transcripts are indicated (tr mito, accurately initiated transcript synthesized by mitochondrial polymerase; tr pol I, accurately initiated pol I-specific transcript). Although equal amounts of nonspecific RNA-synthesizing activities were used (data not shown), transcripts due to mitochondrial polymerase predominated, probably because the Mg2+ and potassium acetate concentration was suboptimal for pol I-dependent transcription. (B Lower) Transcripts generated by mitochondrial RNA polymerase are associated with the template and cannot be cleaved by fraction 41. Run-off transcription with the immobilized template was performed by using the mitochondrial RNA polymerase complex for 20 min. After separating the supernatant (s) and pellet (p), fraction 41 was added (lanes 3 and 4) or omitted (lanes 1 and 2).
To investigate the specificity of the reaction, an experiment was performed in which the same linearized template was simultaneously transcribed by pol I and mitochondrial RNA polymerase. It has been shown that a consensus sequence within the yeast pol I promoter 85 bases upstream of the pol I start site supports promoter-dependent RNA synthesis by mitochondrial RNA polymerase (30). When fraction 41 was added to reactions where transcription is proceeded by both polymerases, only the transcript generated by pol I was shortened (Fig. 3B). Under these experimental conditions all transcripts derived from mitochondrial polymerase remained bound to the template (Fig. 3B). Since the cleavage factor had to distinguish between almost identical DNA/RNA molecules embedded in two different ternary complexes, these results prove both the specificity of the cleavage reaction and the necessity of a ternary complex as prerequisite for the processing activity.
RNA Cleavage Resides in Transcriptionally Active Ternary Complexes and Is Not Restricted to Linear Ends of Transcribed Templates.
If the ternary complexes retain the ability to transcribe after the cleavage event, it should be possible to isolate complexes where pol I is paused on its movement along the template, to subject these purified complexes to the cleavage reaction, and finally to reextend truncated transcripts that are still part of a transcriptionally active complex upon addition of nucleotides. Paused transcription complexes were formed using a 3′-end-extended template lacking cytidine within the first stretch of DNA. When transcription was performed with fraction PA600, a 53-nt RNA fragment was synthesized with a ribonucleotide mixture lacking CTP (Fig. 4, lane 2). Transcription in the presence of all four nucleotides gave rise to a 188-nt run-off transcript (Fig. 4, lane 1). To separate released RNA and to purify formed transcriptionally active complexes, the tailed template was attached to magnetic beads on its 3′ extended end. When the isolated, paused transcription complexes were washed and incubated in the absence of nucleotides but with fraction 41, the RNA was shortened (Fig. 4, lane 3), whereas in the absence of fraction 41 the length of the RNA remained unaffected (Fig. 4, lane 2). After removal of the cleavage factor and addition of ribonucleotides with the exception of CTP, the truncated RNA embedded in a ternary complex was chased to its original size (Fig. 4, lane 4). Supplementing all four nucleotides to the purified complex with the shortened transcript resulted in generation of the full-length run-off fragment (Fig. 4, lane 5). These results indicate that truncated transcripts reside in transcriptional active complexes if the RNA remains associated with the complex and can be reextended. Furthermore, these results rule out that the RNase activity acts only on transcription complexes paused at the ends of linear templates.
Figure 4.
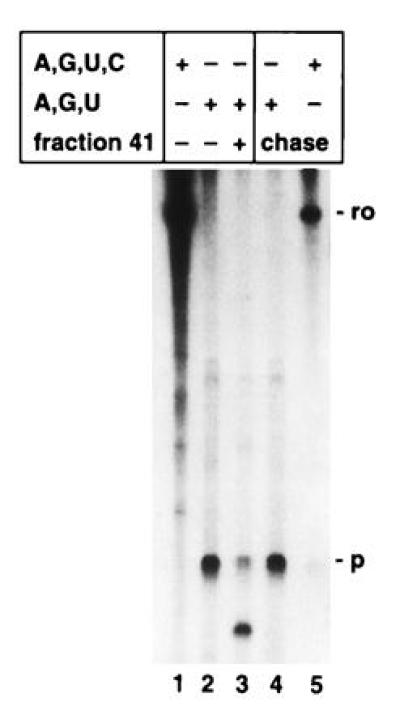
Cleaved transcripts that remain associated within ternary complexes can be reextended upon addition of NTP. Transcription reactions on 3′-extended immobilized templates (pItailKS) were performed as described. In the presence of all four NTPs a 188-nt-long run-off RNA (ro) was generated (lane 1). In the absence of CTP (C), transcription paused at the first CTP to be incorporated (p) and revealed a RNA fragment 53 nt long (lane 2). Purified and washed ternary complexes were incubated without (lane 2) or with (lanes 3–5) fraction 41 for 20 min in the absence of NTPs. After washing the immobilized templates and associated complexes, the complexes were allowed to resume elongation for 20 min at 25°C providing ATP (A), GTP (G), and UTP (U) (lane 4) or all four NTPs (lane 5). RNA fragments were separated on a 7.5% acrylamide/7 M urea gel.
TFIIS Is Not Involved in the Cleavage Reaction.
Since it is known that elongation factor TFIIS promotes cleavage of nascent transcripts generated by pol II, it was necessary to investigate whether this factor plays a similar role in pol I-dependent transcription. This possibility was ruled out by the following observations. When increasing amounts of recombinant yTFIIS (35) were used to substitute for fraction 41 in the pol I-dependent transcription assays, no cleavage could be observed (Fig. 5). Furthermore, TFIIS and the pol I-dependent cleavage activity did not co-chromatograph on the Resource S column. As estimated by Western blotting, TFIIS eluted in a rather broad peak, with its maximum around fraction 30 (data not shown). The pol I-specific cleavage activity eluted in a symmetric peak from fraction 39 to 43. Finally, to rule out that yTFIIS requires other factors present in fraction 41, yTFIIS was immunodepleted from fraction 41 by using an anti-yTFIIS antiserum. Although >95% of yTFIIS could be removed by this procedure, the cleavage efficiency of fraction 41 was not affected (Fig. 5). Taken together, these experiments suggest that yTFIIS is not involved in the described pol I-dependent RNA cleavage.
Figure 5.
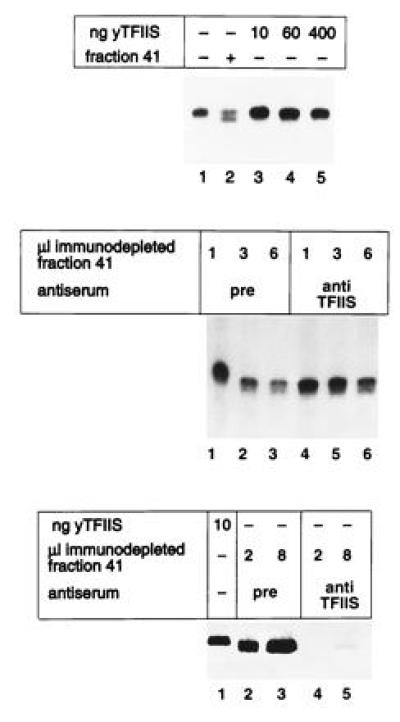
Cleavage reaction is not dependent on TFIIS. (Top) In vitro transcription assays without (lane 1) or with (lane 2) fraction 41 and with increasing amounts of purified recombinant yTFIIS. Recombinant TFIIS was more than 95% pure (generous gift from A. Edwards, Hamilton, Canada). Recombinant TFIIS (5 ng) was able to stimulate pol II-dependent RNA synthesis (data not shown). (Middle) In vitro transcription assays in the presence of fraction 41, which has been immunodepleted by anti-TFIIS antibodies (lanes 4–6) or the corresponding preimmunserum (pre) (lanes 1–3). (Bottom) Immunoblot with antibodies against yTFIIS. Equal amounts of fraction 41 were immunodepleted by an anti-TFIIS antiserum (lanes 4 and 5) and the corresponding preimmunantiserum (lanes 2 and 3). The indicated volumes were subjected to electrophoresis in a 12% SDS/polyacrylamide gel, electroblotted onto nitrocellulose, and probed with anti-TFIIS antibodies. Lane 1 shows 10 ng recombinant purified yTFIIS.
DISCUSSION
None of the pol I-dependent RNase activities described (see Introduction) require the existence of a transcriptionally active ternary complex. So far, the only reported eukaryotic RNase activities dependent on transcriptionally active complexes are associated with pol II/TFIIS and pol III (8, 19). These activities were not responsible for the pol I-dependent transcript cleavage described here. Therefore, the identified pol I-specific RNase activity that was dependent on the presence of a ternary complex is due to a novel protein factor.
In most studies on transcript cleavage reactions in ternary complexes, RNA polymerase paused at intrinsic DNA arrest sites or DNA bound proteins. Elongation arrest can also occur within 10 bases of the end of linear templates, as was shown for pol II-specific transcription (36). The formation of nonchaseable dead ends may explain why E. coli RNA polymerase and pol II stop transcription after traversing certain DNA sequences (5). A similar mechanism could be postulated for pol I. Run-off transcription of immobilized templates demonstrated that a significant amount of pol I-specific transcripts remained bound to the template. So far, no general dependency on the nature of the linearized DNA could be observed: all six differently generated linearized ends supported truncation of at least a detectable amount of transcripts.
At intrinsic blocks of pol II-dependent transcription, TFIIS stimulates read-throughs, permitting elongation, which results in a higher accumulation of the full-length transcripts. Several lines of evidence support the idea that the novel cleavage activity described here might participate in the elongation of pol I-dependent transcription in a similar way. First, in accordance with the TFIIS mediated function, pol I-specific cleavage occurred only if transcription was arrested both at linearized ends and at intrinsic blocks. Second, paused ternary complexes containing truncated transcripts remained transcriptionally active, which is necessary to overcome subsequent transcriptional barriers. However, other observations contradict the direct involvement of the identified RNase activity in RNA chain elongation. (i) Although pol I could be paused by DNA-bound lac repressor and the resulting transcripts could be truncated to some extent under limiting NTP conditions, neither a substantial decrease of the paused transcripts nor an accumulation of the full-length run-off transcripts could be detected when fraction 41 was present during transcription (data not shown). DNA-bound lac repressor seemed not to be an appropriate system to study pol I-specific elongation, since it seems to mimic a termination site to pol I-specific termination (21, 37). Furthermore, when using the enriched pol I initiation complex PA600 for RNA synthesis, most of the transcripts that were stalled at the lac repressor were released from the immobilized template and, therefore, were not accessible for any processing reaction (data not shown). (ii) In contrast to TFIIS and GreA/GreB, the identified RNase activity can be detected even in the presence of nucleotides.
Because of the discrepancies mentioned above, whether the identified activity plays a role in RNA chain elongation still must be clarified. Other functions of a RNA-cleaving activity within ternary complexes should be considered. Although in recent publications on pol I-dependent termination in yeast transcript release was apparently obtained without a special RNA processing activity (38, 39), it is still tempting to consider the participation of a RNA-cleaving factor for transcription termination, especially for the release of a properly terminated transcript. Furthermore, RNA nuclease activity could play an error-correcting role in eukaryotic transcription, a feature that was proposed by several groups (10, 13, 15, 40) and recently suggested to reside in GreA activity during RNA synthesis in E. coli (27).
Acknowledgments
I thank Drs. S. Roeder and N. Lue for plasmid pSES5, Dr. C. Kane for antibodies against yTFIIS, Dr. A. Edwards for recombinant yTFIIS, Drs. H. Bujard and R. Lutz for recombinant lac repressor, and Dr. J. Jaehning for yeast strain yJJ189. Furthermore, I am grateful to Drs. Jennifer Lechner-d’Ortiz, J. Lechner, and E. Hurt for critical reading of the manuscript; to P. Milkereit for helpful discussion; and especially to Dr. I. Haas for advice and thoughtful comments regarding the ongoing project and the manuscript. Above all, I would like to thank Dr. F. Wieland for advice, motivation, and support. The technical assistance of I. Eckstein is gratefully acknowledged. This work was funded by the Deutsche Forschungsgemeinschaft (Ts35/2–1).
Footnotes
Abbreviations: pol I, II, and III, RNA polymerase I, II, and III, respectively; PMSF, phenylmethylsulfonylfluoride.
References
- 1.Surratt C K, Milan S C, Chamberlin M J. Proc Natl Acad Sci USA. 1991;88:7983–7987. doi: 10.1073/pnas.88.18.7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borukhov S, Polyakov A, Nikiforov V, Goldfarb A. Proc Natl Acad Sci USA. 1992;89:8899–8902. doi: 10.1073/pnas.89.19.8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borukhov S, Sagitov V, Goldfarb A. Cell. 1993;72:459–466. doi: 10.1016/0092-8674(93)90121-6. [DOI] [PubMed] [Google Scholar]
- 4.Nudler E, Goldfarb A, Kashlev M. Science. 1994;265:793–796. doi: 10.1126/science.8047884. [DOI] [PubMed] [Google Scholar]
- 5.Arndt K M, Chamberlin M J. J Mol Biol. 1990;213:79–108. doi: 10.1016/S0022-2836(05)80123-8. [DOI] [PubMed] [Google Scholar]
- 6.Nudler E, Kashlev M, Nikiforov V, Goldfarb A. Cell. 1995;81:351–357. doi: 10.1016/0092-8674(95)90388-7. [DOI] [PubMed] [Google Scholar]
- 7.Kane C M. In: Transcription: Mechanisms and Regulation. Conaway R C, Conaway J W, editors. New York: Raven; 1994. pp. 279–296. [Google Scholar]
- 8.Reines D. In: Transcription: Mechanisms and Regulation. Conaway R C, Conaway J W, editors. New York: Raven; 1994. pp. 263–278. [Google Scholar]
- 9.Aso T, Conaway J W, Conaway R C. FASEB J. 1995;9:1419–1428. doi: 10.1096/fasebj.9.14.7589983. [DOI] [PubMed] [Google Scholar]
- 10.Guo H, Price D H. J Biol Chem. 1993;268:18762–18770. [PubMed] [Google Scholar]
- 11.Izban M G, Luse D S. J Biol Chem. 1993;268:12874–12885. [PubMed] [Google Scholar]
- 12.Izban M G, Luse D S. J Biol Chem. 1993;268:12864–12873. [PubMed] [Google Scholar]
- 13.Wang D, Hawley D K. Proc Natl Acad Sci USA. 1993;90:843–847. doi: 10.1073/pnas.90.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reines D, Ghanouni P, Li Q Q, Mote J., Jr J Biol Chem. 1992;267:15516–15522. [PMC free article] [PubMed] [Google Scholar]
- 15.Reines D. J Biol Chem. 1992;267:3795–3800. [PMC free article] [PubMed] [Google Scholar]
- 16.Reines D, Mote J., Jr Proc Natl Acad Sci USA. 1993;90:1917–1921. doi: 10.1073/pnas.90.5.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Izban M G, Luse D S. Genes Dev. 1992;6:1342–1356. doi: 10.1101/gad.6.7.1342. [DOI] [PubMed] [Google Scholar]
- 18.Rudd M D, Izban M G, Luse D S. Proc Natl Acad Sci USA. 1994;91:8057–8061. doi: 10.1073/pnas.91.17.8057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitehall S K, Bardeleben C, Kassavetis G A. J Biol Chem. 1994;269:2299–2306. [PubMed] [Google Scholar]
- 20.Kuhn A, Grummt I. Genes Dev. 1989;3:224–231. doi: 10.1101/gad.3.2.224. [DOI] [PubMed] [Google Scholar]
- 21.Kuhn A, Bartsch I, Grummt I. Nature (London) 1990;344:559–562. doi: 10.1038/344559a0. [DOI] [PubMed] [Google Scholar]
- 22.Huet J, Wyers F, Buhler J-M, Sentenac A, Fromageot P. Nature (London) 1976;261:431–433. doi: 10.1038/261431a0. [DOI] [PubMed] [Google Scholar]
- 23.Iborra F, Huet J, Breant B, Sentenac A, Fromageot P. J Biol Chem. 1978;254:10920–10924. [PubMed] [Google Scholar]
- 24.Marczynski G T, Schultz P W, Jaehning J A. Mol Cell Biol. 1989;9:3193–3202. doi: 10.1128/mcb.9.8.3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stewart S E, Roeder G S. Mol Cell Biol. 1989;9:3464–3472. doi: 10.1128/mcb.9.8.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 27.Erie D A, Hajiseyedjavadi O, Young M C, von Hippel P H. Science. 1993;262:867–873. doi: 10.1126/science.8235608. [DOI] [PubMed] [Google Scholar]
- 28.Sayre M H, Tschochner H, Kornberg R D. J Biol Chem. 1992;267:23376–23382. [PubMed] [Google Scholar]
- 29.Schultz M C, Choe S Y, Reeder R H. Proc Natl Acad Sci USA. 1991;88:1004–1008. doi: 10.1073/pnas.88.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riggs D L, Nomura M. J Biol Chem. 1990;265:7596–7603. [PubMed] [Google Scholar]
- 31.Flanagan P M, Kelleher R J, III, Tschochner H, Sayre M H, Kornberg R D. Proc Natl Acad Sci USA. 1992;89:7659–7663. doi: 10.1073/pnas.89.16.7659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ausubel F M, Brent R, Kingston R E, Moore D M, Seidman J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. New York: Wiley; 1995. [Google Scholar]
- 33.Knittler M R, Haas I G. EMBO J. 1992;11:1573–1581. doi: 10.1002/j.1460-2075.1992.tb05202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lue N F, Kornberg R D. J Biol Chem. 1990;265:18091–18094. [PubMed] [Google Scholar]
- 35.Christie K R, Awrey D E, Edwards A M, Kane C M. J Biol Chem. 1994;269:936–943. [PubMed] [Google Scholar]
- 36.Izban M G, Samkurashvili I, Luse D S. J Biol Chem. 1995;270:2290–2297. doi: 10.1074/jbc.270.5.2290. [DOI] [PubMed] [Google Scholar]
- 37.Jeong S W, Lang W H, Reeder R H. Mol Cell Biol. 1995;15:5929–5936. doi: 10.1128/mcb.15.11.5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lang W H, Morrow B E, Ju Q, Warner J R, Reeder R H. Cell. 1994;79:527–534. doi: 10.1016/0092-8674(94)90261-5. [DOI] [PubMed] [Google Scholar]
- 39.Lang W H, Reeder R H. Proc Natl Acad Sci USA. 1995;92:9781–9785. doi: 10.1073/pnas.92.21.9781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hagler J, Shuman S. J Biol Chem. 1993;268:2166–2173. [PubMed] [Google Scholar]