

Abstract
Previous attempts to express functional DNA cytosine methyltransferase (EC 2.1.1.37) in cells transfected with the available Dnmt cDNAs have met with little or no success. We show that the published Dnmt sequence encodes an amino terminal-truncated protein that is tolerated only at very low levels when stably expressed in embryonic stem cells. Normal expression levels were, however, obtained with constructs containing a continuation of an ORF with a coding capacity of up to 171 amino acids upstream of the previously defined start site. The protein encoded by these constructs comigrated in SDS/PAGE with the endogenous enzyme and restored methylation activity in transfected cells. This was shown by functional rescue of Dnmt mutant embryonic stem cells that contain highly demethylated genomic DNA and fail to differentiate normally. When transfected with the minigene construct, the genomic DNA became remethylated and the cells regained the capacity to form teratomas that displayed a wide variety of differentiated cell types. Our results define an amino-terminal domain of the mammalian MTase that is crucial for stable expression and function in vivo.
Much evidence indicates that in vertebrates the methylation of DNA at cytosine residues affects gene transcription (1). Methylation is believed to be important in a variety of biological processes, including embryonic development, genomic imprinting, and cancer (2, 3). A single gene (Dnmt) encoding a DNA (cytosine-5-)-methyltransferase (MTase; EC 2.1.1.37) has been cloned from mammalian cells (4). MTase shows a marked preference for hemimethylated DNA (5), suggesting that its function is to maintain the methylation status of newly replicated DNA. The enzyme consists of an amino-terminal regulatory domain followed by a 500-amino acid domain assumed to contain the catalytic center, due to its homology to cytosine-methylating bacterial type II restriction methyltransferases (6).
The crucial importance of methylation in development, genomic imprinting, and cancer was demonstrated by the targeted mutation of the Dnmt gene in embryonic stem (ES) cells (7). Mice homozygous for the mutation die at midgestation; homozygous mutant ES cells proliferate normally with their genomic DNA highly demethylated but die upon differentiation (8). Imprinted genes display parent-specific monoallelic expression, and DNA methylation has been proposed to constitute the molecular mark that distinguishes the two alleles (3). Strong support for this notion was provided by the loss of monoallelic expression of these genes in homozygous Dnmt mutant embryos or ES cells (9). Furthermore, a large body of evidence links hypo- and hypermethylation of genomic DNA to cancer progression (10). A direct correlation between DNA methylation and intestinal neoplasia was demonstrated in mice expressing different levels of MTase (11).
The present work used the complementation of the Dnmt mutation as an assay to define the sequences encoding a functional MTase. Previous reports of high expression of functional MTase in transient transfections (12, 13) contrasted with the inability to achieve high expression levels in stable transfection experiments. In fact, only one report described high levels of MTase expression in stably transfected clones that, however, displayed a transformed phenotype (14). We found that the published cDNA was incapable of expressing wild-type levels of MTase and complementing the Dnmt mutation. A construct containing a previously reported (15) Dnmt promoter fused to the Dnmt cDNA also failed in this assay. In contrast, wild-type levels of Dnmt expression were obtained after targeting the published cDNA to its cognate locus, where expression of the full-length cDNA from its cognate promoter fully restored MTase function to ES cells (16). This result suggested that sequences upstream of the previously identified initiation codon were crucial for stable and functional expression of the enzyme. In this paper, we report a previously unidentified amino-terminal ORF that extends the coding capacity of the construct by up to 171 amino acids. Expression of the full-length Dnmt cDNA in mutant ES cells restored both normal methylation levels and the capacity to form teratomas, indicating a crucial role of the amino-terminal domain in normal enzymatic activity.
MATERIALS AND METHODS
Vectors.
Dnmt constructs used were from pMG (T. Bestor, Columbia University, New York), which is a 4934-bp Dnmt cDNA. pMT20 was made by inserting the cDNA into a vector that contains the Pgk-1 promoter and polyadenylylation sequences of ppgk::hprt (17) flanking a synthetic intron (IVS in Fig. 1A) (18). Removal of the IVS from pMT20 yielded pMT10. pMT30 was made by excising a 1357-bp NaeI fragment from pMT10, containing the phage f1 ori, the 3′ end of the lacZ gene, the Pgk-1 promoter, and the first 180 bp of the Dnmt cDNA. A 4.7-kb Dnmt NaeI genomic fragment was cloned into this site, so that the Dnmt cDNA sequence removed from pMT10 was restored in its genomic context. pMT40 was constructed by inserting the Dnmt cDNA sequence into pEF-PGKhyg (a gift of S. Orkin, Howard Hughes Medical Institute, Children’s Hospital, Boston). This variant of pEF-PGKneo (19) included a 1188-bp fragment encompassing the promoter region of the human elongation factor 1α (EF-1α) gene, taken from pEF-BOS (20), a simian virus 40-derived polyadenylylation signal, and a Pgk-1-driven hygR gene. pMT42 was constructed from pMT40 by restriction at the unique NsiI site in the Dnmt cDNA, destruction of the 3′ overhang by T4 DNA polymerase, and insertion of a 12-bp NheI linker (NEB), containing amber stop codons in all three reading frames. pMT50 was made by inserting a 5.0-kb Dnmt EcoRI fragment upstream of the Dnmt genomic region in pMT30. The inserted sequence begins 3641 bp upstream of the first exon and ends 381 bp downstream of the testis-specific exon. The G418-selectable pPGK-RN (21) and the puromycin-selectable pPGK-Puro (16) were made as described.
Figure 1.
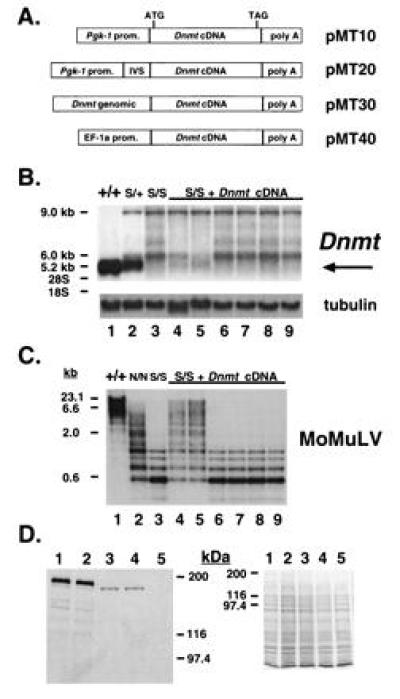
Dnmt cDNA is inefficiently expressed in stable transfections of ES cell lines. (A) Diagram of Dnmt expression constructs. The cDNA used in all four constructs is the same. (B) Northern blot analysis of Dnmt expression in Dnmts/s lines transfected with pMT20 (lanes 4–9). The wild-type Dnmt transcript migrates at 5.2 kb and is indicated by an arrow to the right of the blot. Expression levels are <1% of the wild-type allele in all clones shown, except for lanes 4 and 5, which are 8 and 10%, respectively. Wild-type J1 (lane 1), Dnmts/+ (lane 2), and Dnmts/s (lane 3) cells are shown for comparison. The Dnmt cDNA was used as a probe. The same blot rehybridized with an α-tubulin cDNA (25) to control for amount of RNA loaded is shown also. The bands migrating at 9.0 and 6.0 kb are created by the disruption at the SalI site by a Pgk-Neo cassette and hybridize with a Neo probe (8). (C) Southern blot analysis of HpaII-digested genomic DNA from Dnmts/s lines transfected with pMT20 (lanes 4–9, same order as above). The Moloney murine leukemia virus (MoMuLV) cDNA (26) was used as a probe. Wild-type J1 (lane 1), Dnmtn/n (lane 2), and Dnmts/s (lane 3) cells are shown for comparison. (D) Western blot analysis from Dnmts/s lines transfected with pMT20, which show slight levels of genomic remethylation (lanes 3 and 4 correspond to lanes 4 and 5 in B and C). An amino-terminal anti-MTase antibody was used on the blot to the left and a Coomassie-stained gel is shown as a loading control to the right. Wild-type J1 (lane 1), Dnmts/+ (lane 2), and Dnmts/s (lane 5) cells are shown for comparison. Bio-Rad silver stain molecular weight markers are shown to the side.
Transfections.
The J1 and the Dnmt mutant ES cell lines (7, 8) were cultured as described (7). Each linearized construct was mixed in 5-fold molar excess with 250 ng of selectable marker and the cationic liposome DOTAP (Boehringer Mannheim). Cells were incubated with this mixture according to manufacturer’s protocol and plated on γ-irradiated murine fetal fibroblasts of the appropriate drug resistance. Selection was as follows: G418 (GIBCO/BRL) at 200 μg/ml (active), puromycin (Sigma) at 2 μg/ml, and hygromycin (Boehringer Mannheim) at 100 μg/ml. Isolated colonies were picked 9–12 days after lipofection and expanded. Nucleic acids were analyzed as described (16). Teratomas were made as described (16).
Antibody Derivation and Western Blot Analysis.
A synthetic peptide of the sequence NH2-CRSPRSRPKPRGPRRSK (Mimotopes; Chiron) was coupled to maleimide-activated keyhole limpet hemocyanin (Pierce) and injected into female New Zealand White rabbits (HRP, Denver, PA) with Freund’s complete adjuvant. Booster injections and drawing blood were performed using standard protocols.
Protein was analyzed from clones grown for two passages without feeder cells. Confluent cultures of ES cells in 25-cm2 flasks were lysed in 2× sample buffer followed by boiling for 5 min and sonication. SDS/PAGE on 8% gels were performed according to (22). Western blot analysis was performed according to (23) using antisera HM334 at a 1:3000 dilution and chemiluminescence (ECL, Amersham).
Cloning of 5′ End of Dnmt cDNA.
The first technique used poly(A)+ RNA purified from crude tissue extracts derived from a CD1-mouse kidney using the PolyATract system (Promega). The 5′ RACE system (GIBCO/BRL) was used with 300 ng of poly(A)+ RNA and a Dnmt-specific primer MMT2400AS (5′-AGGGTGTCACTGTCCGACTT, located in the fourth exon). Poly(dC)-tailed cDNA was subjected to 35 PCR cycles using an annealing temperature of 57°C in a 50-μl reaction containing 25 mM Tris·HCl, pH 8.4/50 mM KCl/1.5 mM MgCl2/40 μM of a nested Dnmt-specific primer MMT2363AS (see Fig. 3A)/40 μM of a poly[d(GI)]-anchor primer/10 units of AmpliTaq polymerase (Perkin–Elmer). The PCR product was isolated and cloned into the pCRII vector (Invitrogen).
Figure 3.
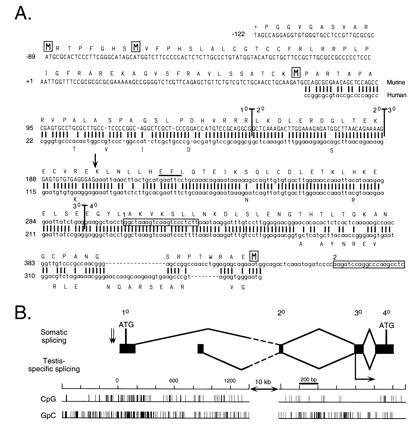
Complete Dnmt cDNA contains two additional 5′ exons that extend the ORF by up to 171 codons. (A) Comparison between murine and human Dnmt sequences at the 5′ end. New sequence reported in this paper is shown in uppercase type and the previously reported sequence is in lowercase type. The first two rows of sequence are genomic sequence, while the five rows beneath represent cDNA sequence. A previously reported human cDNA (31) is shown beneath the murine sequence with all matches represented by a vertical bar. A predicted translation of the 182-codon extension of the ORF is shown above the murine nucleotide sequence, starting with the stop codon TAG and with the four methionine residues boxed. Differences in the human amino acid sequence are represented beneath the human nucleotide sequence. Vertical bars running through the murine sequence denote exon–exon boundaries, with the numbers corresponding to the exon designations used in B. The two boxed sequences represent the primers MMT2214AS (box 1) and MMT2363AS (box 2) used in the cloning experiments. The EcoRI site that marks the beginning of the Dnmt cDNA used in the expression constructs in Fig. 1 is indicated by a bar over the recognition sequence. The major transcriptional start site found by Rouleau et al. (15) is indicated by a vertical arrow. (B) Map of exonic structure of the 5′ end of Dnmt. The first four exons of Dnmt are represented to scale with respect to their size and relative position in the gene. Exon numbers described in text are given above. The splicing of the first four exons of Dnmt is diagrammed above, while the alternative splicing observed in mouse testis (N. B. Kuemmerle, L. E. Halce, and K. Valerie, unpublished work) is shown below. The numbering of the nucleotide scale starts with 0 as the 5′ end of the new cDNA sequence. The two alternative exons are separated from the second common exon by a 10-kb interval of genomic DNA, indicated by the broken lines in the splicing diagram. CpG and GpC incidence diagrams are plotted below to scale with the rest of the diagram. The two 3′-proximal ATGs indicated by a boxed M in A are shown in their genomic locations, while the first two ATGs are indicated by vertical arrows to the left of exon 1. The major transcriptional start site found by Rouleau et al. (15) is indicated by a horizontal arrow below exon 3.
The second method used 400 cm2 of confluent J1 ES cells. RNA was prepared by extraction with guanidine thiocyanate and centrifugation in a cesium chloride step gradient, as described (24). RNA was passed once over an oligo(dT)12–18-cellulose column (Pharmacia) as described (24). Poly(A)+ mRNA (12 μg) was hybridized with 1 μg of MMT2214AS (see Fig. 3A), a Dnmt-specific primer. A cDNA library was constructed using the TimeSaver cDNA synthesis kit (Pharmacia LKB) and ligated with λ ZAP II phage arms (Stratagene). Recombinant phage were packaged and plated out as described (24). Approximately 125,000 clones were screened using a probe containing Dnmt cDNA sequences upstream of the EcoRI site in exon 3 (see Fig. 3A). Standard procedures for hybridization, washing, and autoradiography were observed (24). After a secondary screen, isolated positive clones were picked and converted into plasmids (Stratagene).
The first additional exon (exon 2) was located in the genome by identity to genomic sequence reported by Rouleau et al. (15). The second upstream exon (exon 1) and the testis-specific exon were localized by using the cDNA fragment containing exon 1 as a probe for Southern blots of plasmids containing 129/Sv-derived Dnmt genomic sequences (7).
RESULTS
Dnmt cDNA Is Inefficiently Expressed in Stable Transfections of ES Cells.
To establish stable wild-type and Dnmt mutant cell lines expressing recombinant MTase, four expression constructs carrying the Dnmt cDNA were created (Fig. 1A). The ubiquitously expressed Pgk-1 promoter (27, 28) was used in pMT10 and the human EF-1α promoter (20) was used in pMT40 to direct expression of the Dnmt cDNA. A synthetic intron (18) was inserted into pMT10 to increase gene expression levels (29), creating pMT20. pMT30 was derived from pMT10 by replacing the Pgk-1 promoter and the first 180 bp of the Dnmt cDNA with a 4.7-kb Dnmt genomic fragment (see Fig. 4A) containing a previously reported Dnmt promoter (15) and the excised 180 bp of cDNA, restored in its genomic context.
Figure 4.
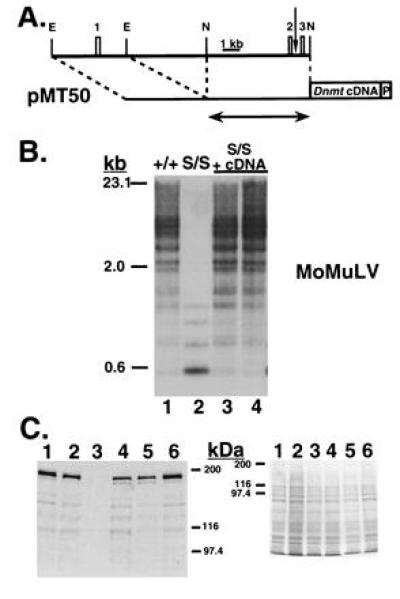
Restoration of normal genomic DNA methylation levels upon expression of the full-length Dnmt cDNA. (A) Map of pMT50, showing its relation to Dnmt genomic sequence and the exons therein (drawn to scale). The first three exons of Dnmt are represented by numbered boxes and correspond to those shown in Fig. 3B. The vertical arrow represents the transcriptional start site reported in Rouleau et al. (15). EcoRI (E) and NaeI (N) sites used in cloning are indicated. The Dnmt cDNA and Pgk-1 polyadenylylation signal (P) are both represented by boxes. The amount of Dnmt genomic sequence used in pMT30 is shown beneath by the arrow. (B) Southern blot analysis of HpaII-digested genomic DNA, probed with the MoMuLV cDNA. Wild-type J1 (lane 1) and Dnmts/s cells (lane 2) are shown, followed by pMT50-transfected Dnmts/s cells (lanes 3 and 4). (C) Western blot analysis of the pMT50-transfected cells shown in B (lanes 4 and 5). An amino-terminal anti-MTase antibody was used in the blot on the left and a Coomassie-stained gel is shown as a loading control on the right. Wild-type J1 (lanes 1 and 6), Dnmts/+ (lane 2), and Dnmts/s (lane 3) cells are shown for comparison. Bio-Rad silver stain molecular weight markers are shown to the side.
The four constructs were separately cotransfected into wild type Dnmt J1 ES cells (7) and the homozygous mutant Dnmts/s (8) and Dnmtn/n cell lines (7). The Dnmts allele is completely inactivated whereas the Dnmtn allele is a partial loss-of-function mutation. Cells bearing either mutation in a homozygous state stably maintain a low level of DNA methylation (7, 8) (Fig. 1C, lanes 2 and 3). Drug-resistant colonies were picked after 9–12 days of selection. Genomic DNA was extracted and examined by Southern blot analysis using Dnmt genomic and cDNA probes, which revealed high-copy-number integrants with no gross rearrangements of the constructs (data not shown). Eighteen positive clones were analyzed for mRNA expression by Northern blot analysis, using the Dnmt cDNA as a probe. The size of the endogenous mRNA is 5.2 kb (Fig. 1B, lane 1), while the expected size from the transfected expression constructs was 5.1 kb. In the Dnmts/s ES cells (lane 3), no 5.2-kb transcript was present, although aberrant transcripts of 9.0 and 6.0 kb representing abnormal splicing events caused by the insertion of the neomycin-resistance gene were observed (8). Fig. 1B (lanes 4–9) shows a low level of expression in one-third of the Dnmts/s clones transfected by pMT20 (lanes 4 and 5), while the other transfectants (lanes 6–9) displayed no detectable expression. The decrease of the aberrant 6.0-kb transcript in clones expressing low levels of pMT20 cannot be explained at present (lanes 4 and 5). Quantitation of band intensities using α-tubulin as a loading control (Fig. 1B) revealed that, in 5 of 18 clones, Dnmt cDNA expression levels were 5–10% of wild-type Dnmt, whereas the remaining clones showed no detectable cDNA expression. Similarly, 23 J1 clones transfected with the same constructs showed no significant increase in levels of Dnmt expression over untransfected J1 ES cells (data not shown).
To investigate whether the low Dnmt cDNA expression levels would result in an increase in global DNA methylation, Southern blot analysis of genomic DNA digested with HpaII, an enzyme sensitive to the methylation of internal cytosines in its CCGG recognition sequence, was performed. A MoMuLV cDNA probe that cross-hybridizes with endogenous C-type retroviruses (26) that are heavily methylated in wild-type cells was used (Fig. 1C, lane 1). DNA methylation in Dnmt mutant cells is greatly reduced, as reflected by the increasing intensity of lower molecular weight bands in this assay, with DNA methylation levels in the Dnmts/s line (lane 3) lower than in the Dnmtn/n line (lane 2). A slight increase in genomic methylation was detected only in those Dnmts/s clones that had been transfected by either pMT20 (Fig. 1C, lanes 4 and 5) or pMT40 (data not shown) and showed plasmid expression of >5% of the wild-type allele. The extent of remethylation was slightly higher than that seen in untransfected Dnmtn/n cell lines. By this assay, 8 of 19 Dnmts/s clones containing a Dnmt cDNA displayed partial genomic remethylation, and this methylation level was stably maintained through five passages (data not shown). Also, inclusion into the expression construct pMT30 of the upstream genomic Dnmt sequences that had previously been reported to drive transcription (15) did not lead to genomic remethylation in Dnmtn/n cells (0 of 24 clones).
Western blot analysis, using an antibody directed against the amino terminus of the protein, indicated levels of MTase approximately 5–10% that of wild type in pMT20-transfected clones (Fig. 1D). Moreover, this recombinant MTase (lanes 3 and 4) was shorter by approximately 8 kDa than the protein seen in J1 cells (lane 1). We conclude that, using either heterologous promoters or the reported cognate Dnmt promoter (15), only low levels of the Dnmt cDNA are expressed in stably transfected wild-type or mutant ES cells.
Dnmt cDNA Is Efficiently Expressed From Carboxyl-Terminal Truncated Constructs.
To test the hypothesis that the low mRNA expression was caused by the protein product of the Dnmt expression constructs being detrimental to cells, a construct was designed such that translation of the Dnmt cDNA would end prematurely, producing truncated and possibly inactive peptides. A stop codon-containing linker was inserted into pMT40 at the unique NsiI site 871 bp downstream of the initial ATG in the Dnmt cDNA, to create pMT42 (Fig. 2). This site is located in the middle of the domain that targets MTase to sites of DNA synthesis in S-phase nuclei (30). Translation of this message would be expected to produce a truncated protein of 292 amino acids with neither catalytic activity nor the ability to colocalize with replication foci. Dnmts/s ES cells were transfected and individual colonies were examined by Northern blot analysis. In contrast to a typical clone carrying the wild-type cDNA (Fig. 2, lane 4), high levels of RNA were produced in 4 of 8 clones carrying the mutant cDNA construct (Fig. 2, lanes 7, 8, 11, and 12). As expected, none of the transfectants showed genomic remethylation (data not shown). This conclusion was corroborated by a reduced number of clones obtained with pMT40 (containing the wild-type ORF) as compared with pMT42 (carrying the stop codon): transfection of 5 × 106 ES cells with the latter clone yielded 500 colonies compared with only 200 seen with pMT40. These results are consistent with a detrimental effect of the protein produced from the original expression constructs resulting in selection against cells expressing high levels of the truncated Dnmt cDNA.
Figure 2.

Dnmt cDNA is efficiently expressed from carboxyl-terminal truncated constructs. (Upper) pMT42, made by inserting a stop-codon containing linker into the NsiI site in pMT40 871 bp downstream of the initial ATG. (Lower) Northern blot analysis of Dnmt expression in Dnmts/s lines transfected with pMT42. Four of eight of the examined transfectants (lanes 5–12) showed an expression level approximately 50% that of the wild-type cells. The following lines are shown for comparison: wild-type J1 (lane 1), Dnmts/+ (lane 2), Dnmts/s (lane 3), and D23 (lane 4), a pMT40-transfected Dnmts/s cell line showing slight levels of Dnmt cDNA expression and genomic remethylation. The Dnmt cDNA was used as a probe. At the bottom is shown the same blot rehybridized with an α-tubulin cDNA to control for amount of RNA loaded. S allele-specific bands are as described in Fig. 1B.
The Complete Dnmt cDNA Contains Two Additional 5′ Exons That Extend the Open Reading Frame by at Least 118 Codons.
The results described above suggested that high expression of the Dnmt cDNA was not tolerated in transfected cells. Given the results of Western blot analysis (Fig. 1D), it appeared that the cDNA was incomplete, encoding a truncated protein that was toxic to cells. Therefore, we attempted to derive further upstream sequences. First, CD1 kidney mRNA was used in 5′-RACE experiments that employed the Dnmt-specific primer MMT2400AS for first-strand cDNA synthesis. Subsequent PCR amplification of the cDNA product with the Dnmt-specific primer MMT2363AS (Fig. 3A) yielded a product that included two additional exons.
Independently, poly(A)+ mRNA from J1 ES cells was used for the construction of a cDNA library. The Dnmt-specific primer MMT2214AS (Fig. 3A) was employed for first-strand synthesis, and 105 phage clones were screened with a probe containing Dnmt cDNA sequences upstream of the EcoRI site in the third exon (Fig. 3A). Examination of 12 clones revealed the same sequence cloned by 5′-RACE, with the longest of the clones extending the 5′-RACE product by 34 bp. In contrast to previous reports of Dnmt sequence (4, 15), close examination identified a 141-codon extension of the published ORF, including a new initial ATG 118 codons upstream of the previously published initial ATG (Fig. 3A).
Comparison with the published Dnmt genomic DNA sequence (15) revealed that the new cDNA sequence contains a 15-bp 5′ extension of the published first exon (exon 3) and two new exons (exons 2 and 1, respectively; Fig. 3B). The 37-bp exon 2 is separated from the 150-bp exon 3 by a 767-bp intron, while exon 1 lies 11.5 kb upstream of exon 2. Exon 1 was found in the center of a CpG island (Fig. 3B) and hence provides the Dnmt gene with the typical elements of a housekeeping gene. Examination of the genomic sequence around the new primary exon revealed a continuation of the ORF for another 122 bp upstream of the 5′ limit of the current cDNA sequence. This additional genomic sequence begins with an in-frame stop codon and contains two in-frame ATGs (Fig. 3A). An alternatively spliced upstream exon isolated both from mouse testis and HeLa cells (N. B. Kuemmerle, L. E. Halce, and K. Valerie, unpublished work) (EMBL accession no. X77486X77486) was found to lie 800 bp downstream of exon 1 (Fig. 3B). These results show that tissue-specific and alternative transcriptional start sites in mouse and human tissues are localized upstream of the previously characterized 5′ end of the gene (15).
A comparison of the new murine amino-terminal cDNA sequence with the published human cDNA (31) showed that these sequences are highly conserved, especially at the beginning of the ORF (Fig. 3A). Similar to the murine sequence, the human sequence contains an ORF upstream of the published ATG initiation codon. In the first shared 282 bp, the respective gene sequences are 85% identical at the nucleotide level, and 91% identical at the amino acid level. Thirty-three of 41 nucleotide mismatches between the two sequences result in silent mutations. This evolutionary conservation may indicate an important and essential function of this amino-terminal sequence, consistent with the data presented here.
Expression of Complete Dnmt cDNA in Dnmt Mutant ES Cells Restores Normal Genomic Methylation Levels.
To test the possibility that the newly discovered cDNA sequence would allow normal expression and MTase functioning in vivo, a minigene expression construct was made and stably transfected into Dnmt mutant ES cells. pMT50 was made by insertion of a 5.0-kb region of the Dnmt locus that included the first exon into pMT30 (Fig. 4A). pMT30 contained the previously reported Dnmt promoter (15) but had been shown not to function in Dnmt mutant ES cells (Fig. 1A). pMT50 resulted in the Dnmt cDNA fused 3′ to a 9.7-kb piece of genomic Dnmt sequence that ended in the fourth exon and included 3.6 kb of genomic sequence upstream of the first exon, the two new exons reported above, three introns, and the beginning of the fourth exon.
Dnmts/s cells were electroporated with the linearized construct and selected for puromycin resistance. Puromycin-resistant colonies were expanded and examined by Southern blot analysis for vector integration and levels of genomic methylation. Seven of 12 clones examined showed random integration into the genome (data not shown) and normal levels of genomic methylation (Fig. 4B), as compared with wild-type cells. Two of 12 clones had undergone an homologous targeting of the construct to the endogenous Dnmt locus and the remaining 3 clones had no transgene (data not shown). All clones carrying an intact transgene displayed equivalent methylation profiles at repetitive gene sequences and Xist. The imprinted genes H19 and Igf2r, however, were not remethylated (data not shown), as reported previously for a smaller similar Dnmt cDNA-containing plasmid that was targeted to the endogenous Dnmt locus (16). Western blot analysis revealed a range of MTase levels from 10–100% that of a wild-type allele, with the minigene-encoded enzyme comigrating with wild-type MTase (Fig. 4C).
Three of the rescued lines were used to induce teratomas by subcutaneous injection into syngeneic male host animals. While the parental mutant cells failed to form palpable teratomas within 3–4 weeks (n = 0/12), the rescued ES cells induced teratoma formation at a rate (n = 9/10) similar to the rate (n = 6/6) of wild-type J1 ES cells. Histological analysis of these teratomas revealed a wide variety of differentiated cell types, consisting mostly of mature neural tissue and including discrete patches of epithelial tissue, cartilage, trophectoderm, osteoid, and a relative deficiency of striated muscle (data not shown). From these results we conclude that the additional sequences uncovered above are necessary for functional activity of MTase in vivo.
DISCUSSION
The experiments in this study were prompted by the failure of published Dnmt cDNA clones to functionally complement the hypomethylation phenotype of ES cells homozygous for various mutations of the Dnmt gene. Our results allow two main conclusions regarding the structure and function of DNA methyltransferase. (i) Stably transfected ES cells failed to express high levels of the previously described Dnmt cDNA clone, whereas expression of the full-length cDNA led to wild-type expression levels. We conclude that the previously described cDNA is truncated, lacking an ORF of up to 171 codons upstream of the ATG that was previously proposed to represent the initiation codon (4). (ii) Genomic methylation levels in ES cells, which were decreased substantially as a result of the targeted inactivation of the Dnmt gene, were restored to normal levels by expression of the Dnmt minigene. This restoration of methylation was accompanied by functional rescue of the transfected cells, as reflected in the ability to form teratomas. Rescued cells behave identically to those reported (16) in which the Dnmt cDNA was targeted to its cognate locus. We conclude that the inclusion of the newly identified sequence is essential for stable expression and methyltransferase activity in vivo.
Functional rescue of the Dnmt mutant phenotype suggests that normal methylation patterns can by restored to assure appropriate gene expression in differentiating ES cells. De novo methylation of the demethylated genome is likely accomplished by an independently encoded DNA methyltransferase, as discussed elsewhere (8, 16). Our results argue that demethylation of the mutant ES cell genome does not result in irreversible alteration in gene activity and/or chromatin structure. Rescue of the mutant phenotype seems to be the result of a default de novo methylation process that requires the expression of the hemimethyltransferase encoded by Dnmt that would maintain methylation levels set by the embryonic de novo methyltransferase.
Both the low expression levels in cells carrying the truncated cDNA in stably transfected ES cells and the restoration of high levels of expression from constructs containing an in-frame stop codon in the ORF were consistent with a truncated polypeptide being produced from the cDNA, translation of which may be detrimental to ES cells. Indeed, we isolated additional upstream cDNA sequence containing two exons separated by an 11.5-kb intron. Examination of the genomic sequence surrounding this new primary exon revealed an uninterrupted ORF that begins upstream of and continues throughout the two new exons, extending the published coding sequence by 171 codons. Three new in-frame ATG codons were found in this ORF. Lack of these extra sequences could account for the size differences between MTase purified from cell lines and the product expressed from the truncated Dnmt cDNA (12). Comparison of the mobility in SDS/PAGE of wild-type MTase with the recombinant MTase produced from the expression constructs (Figs. 1D and 4C) suggests the third ATG in the ORF most likely represents the translational start site. Although the results do not determine which of the three upstream ATGs is used as a translational start point in vivo, they do show that the new ORF is necessary for proper functioning in vivo. Protein sequencing will resolve which ATG is used as a translational start.
The first exon was found to lie within a CpG island, as defined by Bird (32). Because of the G+C-rich nature of the sequence, it is difficult to ascertain whether the 5′-end heterogeneity of the 12 examined clones reflects pausing by the reverse transcriptase or a multiplicity of transcriptional start sites, as often observed in housekeeping promoters associated with a CpG island. Our results are not in agreement with the position of the Dnmt promoter localized upstream of exon 3, as reported by Rouleau et al. (15). Expression from this promoter would produce a truncated MTase that is detrimental to stably transfected cells, and Dnmt expression constructs utilizing this promoter failed to effect complementation in Dnmt mutant cells. Instead we suggest that a promoter contained within the pMT50 construct represents the functionally relevant transcriptional start site.
Several reports have demonstrated the ability to express high levels of the truncated Dnmt cDNA in a transient fashion (12, 13). In contrast, only a few reports have described high levels of MTase expression in stably transfected clones of transformed cells (14, 33) or in a myoblast cell line (25). This raises the question of whether specific functional domains of the polypeptide may explain the low Dnmt expression and the apparent toxicity of the truncated cDNA when stably expressed in ES cells. It has been shown previously that the truncated Dnmt cDNA, when expressed in COS cells, generates an enzyme that methylates a hemimethylated DNA substrate with the same kinetics as MTase purified from MEL cells (12). Proteolytic cleavage of the first 350 amino acids did not affect in vitro enzyme activity (34). Consistent with these observations, expression of the truncated Dnmt cDNA resulted in limited genomic remethylation in Dnmts/s ES cells, raising the possibility that the detrimental effect of truncated MTase in ES cells can be separated from its methylating function. MTase is localized to nuclear DNA replication sites during S phase by an amino-terminal targeting domain (30). Because insertion of stop codons within this targeting domain abolished the toxic effect of truncated cDNA expression, we speculate that toxicity may be mediated through this domain, possibly by targeting the truncated MTase to replication sites and thereby stalling the replication machinery. The isolation of a complete Dnmt cDNA allows for analysis of these and many other biological questions, which can finally be addressed through overexpression and tissue-specific expression of the enzyme. The identification of a new transcriptional start site also reopens the discussion on the regulation of Dnmt gene expression, which is known to be complex.
Note Added in Proof.
A new 5′ ORF in the Dnmt gene has also been found by others (see ref. 35).
Acknowledgments
We thank Tim Bestor for pMG and Stuart Orkin for pEF-PGKhyg. K.T. was supported by National Cancer Institute Training Grant T32-CA09541. D.T. was supported by the Human Frontier Science Program. H.L. was supported by the Council for Tobacco Research. R.J. was supported by the National Institutes of Health Grant R35-CA44339.
Footnotes
References
- 1.Yeivin A, Razin A. In: DNA Methylation: Molecular Biology and Biological Significance. Jost J P, Saluz H P, editors. Basel: Birkhauser; 1993. pp. 533–568. [Google Scholar]
- 2.Jones P A, Rideout W M, Shen J C, Spruck C H, Tsai Y C. BioEssays. 1992;82:9–12. doi: 10.1002/bies.950140107. [DOI] [PubMed] [Google Scholar]
- 3.Barlow D P. Science. 1995;270:1610–1613. doi: 10.1126/science.270.5242.1610. [DOI] [PubMed] [Google Scholar]
- 4.Bestor T, Laudano A, Mattaliano R, Ingram V. J Mol Biol. 1988;203:971–983. doi: 10.1016/0022-2836(88)90122-2. [DOI] [PubMed] [Google Scholar]
- 5.Gruenbaum Y, Cedar H, Razin A. Nature (London) 1982;295:620–622. doi: 10.1038/295620a0. [DOI] [PubMed] [Google Scholar]
- 6.Leonhardt H, Bestor T H. In: DNA Methylation: Molecular Biology and Biological Significance. Jost J P, Saluz H P, editors. Basel: Birkhauser; 1993. pp. 109–119. [Google Scholar]
- 7.Li E, Bestor T H, Jaenisch R. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 8.Lei H, Oh S P, Okano M, Jüttermann R, Goss K A, Jaenisch R, Li E. Development (Cambridge, UK) 1996;122:3195–3205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]
- 9.Li E, Beard C, Jaenisch R. Nature (London) 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- 10.Laird P W, Jaenisch R. Hum Mol Genet. 1994;3:1487–1495. doi: 10.1093/hmg/3.suppl_1.1487. [DOI] [PubMed] [Google Scholar]
- 11.Laird P W, Jackson-Grusby L, Fazeli A, Dickinson S, Jung W E, Li E, Weinberg R A, Jaenisch R. Cell. 1995;81:197–205. doi: 10.1016/0092-8674(95)90329-1. [DOI] [PubMed] [Google Scholar]
- 12.Czank A, Hauselmann R, Page A W, Leonhardt H, Bestor T H, Schaffner W, Hergersberg M. Gene. 1991;109:259–263. doi: 10.1016/0378-1119(91)90618-l. [DOI] [PubMed] [Google Scholar]
- 13.Glickman J F, Reich N O. Biochem Biophys Res Commun. 1994;204:1003–1008. doi: 10.1006/bbrc.1994.2562. [DOI] [PubMed] [Google Scholar]
- 14.Wu J, Issa J P, Herman J, Bassett D E, Jr, Nelkin B D, Baylin S B. Proc Natl Acad Sci USA. 1993;90:8891–8895. doi: 10.1073/pnas.90.19.8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rouleau J, Tanigawa G, Szyf M. J Biol Chem. 1992;267:7368–7377. [PubMed] [Google Scholar]
- 16.Tucker K L, Beard C, Dausman J, Jackson-Grusby L, Laird P W, Lei H, Li E, Jaenisch R. Genes Dev. 1996;10:10008–10020. doi: 10.1101/gad.10.8.1008. [DOI] [PubMed] [Google Scholar]
- 17.van der Lugt N, Maandag E R, te Riele H, Laird P W, Berns A. Gene. 1991;105:263–267. doi: 10.1016/0378-1119(91)90161-4. [DOI] [PubMed] [Google Scholar]
- 18.Huang M, Gorman C. Nucleic Acids Res. 1990;18:937–947. doi: 10.1093/nar/18.4.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhen L, King A A J, Xiao Y, Chanock S J, Orkin S H, Dinauer M C. Proc Natl Acad Sci USA. 1993;90:9832–9836. doi: 10.1073/pnas.90.21.9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mizushima S, Nagata S. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rudnicki M A, Braun T, Hinuma S, Jaenisch R. Cell. 1992;71:383–390. doi: 10.1016/0092-8674(92)90508-a. [DOI] [PubMed] [Google Scholar]
- 22.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 23.Vassalli A, Matzuk M M, Gardner H A R, Lee K-F, Jaenisch R. Genes Dev. 1994;8:414–427. doi: 10.1101/gad.8.4.414. [DOI] [PubMed] [Google Scholar]
- 24.Maniatis T, Fritsch E, Sambrook J. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 25.Takagi H, Tajima S, Asano A. Eur J Biochem. 1995;231:282–291. doi: 10.1111/j.1432-1033.1995.tb20698.x. [DOI] [PubMed] [Google Scholar]
- 26.Stuhlmann H, Jahner D, Jaenisch R. Cell. 1981;26:221–232. doi: 10.1016/0092-8674(81)90305-6. [DOI] [PubMed] [Google Scholar]
- 27.McBurney M W, Sutherland A C, Adra C N, Leclair B, Rudnicki M A, Jardine K. Nucleic Acids Res. 1991;19:5755–5761. doi: 10.1093/nar/19.20.5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McBurney M W, Staines W A, Boekelheide K, Parry D, Jardine K, Pickavance L. Dev Dyn. 1994;200:278–293. doi: 10.1002/aja.1002000403. [DOI] [PubMed] [Google Scholar]
- 29.Choi T, Huang M, Gorman C, Jaenisch R. Mol Cell Biol. 1991;11:3070–3074. doi: 10.1128/mcb.11.6.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leonhardt H, Page A W, Weier H U, Bestor T H. Cell. 1992;71:865–873. doi: 10.1016/0092-8674(92)90561-p. [DOI] [PubMed] [Google Scholar]
- 31.Yen R W, Vertino P M, Nelkin B D, Yu J J, el-Deiry W, Cumaraswamy A, Lennon G G, Trask B J, Celano P, Baylin S B. Nucleic Acids Res. 1992;20:2287–2291. doi: 10.1093/nar/20.9.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bird A P. Nature (London) 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 33.Vertino P M, Yen R-W C, Gao J, Baylin S B. Mol Cell Biol. 1966;16:4555–4565. doi: 10.1128/mcb.16.8.4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bestor T H, Ingram V M. Proc Natl Acad Sci USA. 1985;82:2674–2678. doi: 10.1073/pnas.82.9.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoder, J. A., Yen, R.-W. C., Vertino, P. M., Bestor, T. H. & Baylin, S. B. (1996) J. Biol. Chem., in press. [DOI] [PubMed]