

Abstract
Using a novel Escherichia coli in vitro decay system in which polysomes are the source of both enzymes and mRNA, we demonstrate a requirement for poly(A) polymerase I (PAP I) in mRNA turnover. The in vitro decay of two different mRNAs (trxA and lpp) is triggered by the addition of ATP only when polysomes are prepared from a strain carrying the wild-type gene for PAP I (pcnB+). The relative decay rates of these two messages are similar in vitro and in vivo. Poly(A) tails are formed on both mRNAs, but no poly(A) tails are detected on the 3′ end of mature 23S rRNA. The size distribution of poly(A) tails generated in vitro, averaging 50 nt in length, is comparable to that previously reported in vivo. PAP I activity is associated exclusively with the polysomes. Exogenously added PAP I does not restore mRNA decay to PAP I− polysomes, suggesting that, in vivo, PAP I may be part of a multiprotein complex. The potential of this in vitro system for analyzing mRNA decay in E. coli is discussed.
In Escherichia coli, mRNA decay has been studied extensively in vivo with a variety of approaches. The identification of exo- and endoribonucleases and the use of mutant forms of these enzymes (1–5) has resulted in the recognition of decay intermediates in vivo (6–9) and hypotheses to explain the temporal process of decay for several messages (9–11). In addition, careful analysis of some messages has demonstrated physical characteristics of both the 5′- and 3′-untranslated regions that profoundly affect the accessibility of these mRNAs to ribonucleolytic attack (12–15). More recent evidence has indicated that factors other than ribonucleases and RNA structure may modulate message decay, particularly the role of translating ribosomes (16) and the activity of poly(A) polymerase (PAP) I (17, 18).
Although our understanding of mRNA decay has progressed considerably, many questions remain regarding the overall biochemical mechanism. In other complex biological pathways, such as DNA replication and protein synthesis, in vitro studies have provided invaluable assistance for isolating and characterizing important system components. In the case of mRNA decay, however, no useful in vitro system has been described. To be of value, an in vitro reaction should accurately reflect the relative decay rates of short- and long-lived messages (19) and generate the same breakdown products observed in vivo. The use of labeled run-off mRNA transcripts in the presence of E. coli extracts has not met these criteria (unpublished results).
As a first step in developing a reliable in vitro system, we present studies using mRNAs associated with polysomes as substrates for decay. This approach was adopted because in vivo decay has been shown to be affected by translation rates (16, 20). In addition, we hypothesized that many of the factors necessary for mRNA decay might be associated with polyribosomes. Because of the complexity of such a system, we chose to focus initially on whether polyadenylylation affected mRNA decay rates in vitro.
PAP activity was first identified in E. coli in 1962 (21) and was shown subsequently to be associated with the ribosomes (22). However, because no functional role for the enzyme was found, this association was assumed to be fortuitous (22). The identification of pcnB (for plasmid copy number) as the structural gene for PAP I (23) has made it possible to examine the role of polyadenylylation in mRNA metabolism. Several laboratories have now shown that polyadenylylation is involved in mRNA decay in vivo (17, 18) and in the breakdown of a structural RNA, namely RNA I of plasmid ColE1 (24, 25). One recently outlined model for mRNA decay in E. coli suggests that a poly(A) tail targets an mRNA for decay (17, 26). A prediction of this model is that polyadenylylation should stimulate mRNA decay in vitro if the system were appropriately constituted.
The data presented here confirm the association of PAP I with polysomes and demonstrate that, in vitro, polyadenylylation triggers the degradation of both the polysome-associated lpp and trxA mRNAs. The relative decay rates of these two messages in vitro are comparable with the rates observed in vivo. The in vitro decay of the lpp and trxA messages was dramatically reduced when polysomes were isolated from a PAP I− (ΔpcnB) strain. In addition, the poly(A) tails generated in vitro were approximately the same length as those in vivo. Using site-specific digestion with RNase H, it was also demonstrated that polyadenylylation occurs on mRNA species and not at the mature 3′ end of 23S ribosomal RNA. Our results are discussed in relation to the role of poly(A) tailing in message decay and the potential of this approach for developing a complete in vitro mRNA decay system.
MATERIALS AND METHODS
Bacterial Strains.
All strains were derived from E. coli MG1693 (thyA715; provided by B. Bachmann, E. coli Genetic Stock Center, Yale University) and contained a null mutation of RNase I (rna-19; ref. 27), to avoid nonspecific degradation of ribosomes. Strains were constructed using P1-mediated transduction (28). SK5667 (pcnB+ rna-19 thyA715; designated in text as PAP I+) was obtained based on the cotransduction of rna and leuS. The temperature-sensitive leuS31 allele was transduced into MG1693 by selecting for a 66% linked Tn10 (Tcr) insertion. A Tcr transductant that could not grow on minimal medium at 42°C was purified and used as a recipient in a cross with P1 grown on SK2299 (argG6 hisG1 metB1 rpsL104 rna-19). Transductants able to grow at 42°C in the absence of leucine were tested for RNase I deficiency using the methyl green test (ref. 29; S. D. Yancey and S.R.K., unpublished results). SK8964 (ΔpcnB::kan rna-19 thyA715; designated in text as PAP I−) was derived from SK5667 using a kanamycin resistance (Kmr) selection for the ΔpcnB::kan deletion/substitution mutation (30). SK8317 (caa::cam ΔpcnB::kan rna-19 thyA715) was constructed by transducing SK8964 (ΔpcnB::kan rna-19 thyA715) with bacteriophage P1 grown on CA244 (caa::cam; provided by M. Deutscher, University of Miami, Miami) and selecting for Cmr transductants. The plasmid pJL89 (30) is a low-copy number plasmid carrying pcnB+.
Polysome Preparation.
Polysomes were prepared using the procedure of Ron et al. (31) from cultures grown in Luria broth (32) at 37°C to an OD595 of 0.5. The 30,000 × g cleared extract (S30) was layered over 4 ml of 30% sucrose and centrifuged in a Beckman SW40 rotor at 35,000 rpm for 90 min. The polysome pellet was resuspended in 10 mM Tris, pH 7.4/10 mM MgOAc/2 mM DTT and kept on ice. Absorption at OD260 was used to quantify the preparations. Protein concentrations were determined using the Bio-Rad protein assay.
Enzymes and Reagents.
Nucleotides, RNase H, RNase T1, RNase A, and T4 RNA ligase were purchased from Boehringer Mannheim. Radionucleotides were obtained from NEN, and PAP I was purchased from Bio-Rad. Biotrans+ membrane (ICN) was used for all Northern blots. Catrimox-14, a cationic surfactant solution, was obtained from Iowa Biotechnology (Coralville, IA).
PAP Activity.
PAP activity was assayed as the incorporation of [32P]AMP into RNA. Samples were removed at various intervals from reaction mixtures used for in vitro mRNA decay to which 1 μl of [α-32P]ATP (3000 Ci/mmol; 1 Ci = 37 GBq) was added. The reactions were stopped by the addition of 10 mM EDTA and 1% SDS. After dilution to 100 μl with deionized H2O, an equal volume of Catrimox-14 was added. The resulting precipitated protein, DNA, and RNA were pelleted by centrifugation at room temperature. The pellet was resuspended in 1 ml of 2 M LiCl, in which the protein and DNA were soluble but the RNA was not. Following centrifugation, the pelleted RNA was washed twice with 70% ethanol, dried, and counted in Eppendorf tubes by the Cherenkov method (33). This method was more sensitive than precipitation with trichloroacetic acid and permitted subsequent determination of both poly(A) tail length on 12% polyacrylamide gels and the distribution of incorporated label on 6% denaturing polyacrylamide gels.
In Vitro mRNA Decay Assay.
A 15-μl reaction mixture containing 0.25 OD260 units of polysome preparation in 50 mM Tris (pH 8)/10 mM MgCl2/4 mM MnCl2/150 mM NaCl/2 mM DTT/1 mM EDTA/15 μg of acetylated BSA and 50 μM ATP was incubated at 37°C. The reaction was stopped by the addition of 10 mM EDTA and 1% SDS. The samples, diluted to 100 μl with deionized H2O to give a final concentration of 0.2% SDS, were extracted twice with phenol (pH 5.2) and precipitated with 0.1 volume of 2 M NaOAc and 2.5 volumes of ethanol. Northern blots were prepared and assayed as described by O’Hara et al. (17). Decay rates were determined from the slope of the logarithmic plot of the percent signal remaining versus time using data from at least three independent polysome preparations.
In Vivo mRNA Decay Assay.
Total RNA was prepared from cultures grown in Luria broth at 37°C to an OD595 of 0.5. One minute after the addition of nalidixic acid (20 μg/ml) and rifampicin (500 μg/ml, dissolved in dimethyl sulfoxide), the first sample, representing time zero, was taken. Subsequent samples were removed at the same time intervals as for the in vitro assay. RNA samples were isolated and analyzed as described by O’Hara et al. (17).
RNase H Digestion.
Site-specific mRNA cleavage was performed using gene-specific antisense oligonucleotides and digestion with RNase H according to the method of Donis-Keller (34). Deadenylylation of mRNA was achieved by RNase H digestion in the presence of oligo(dT)20, as described by Narayan et al. (35).
Determination of Poly(A) Tail Length.
Poly(A) tail lengths were determined by either adding [α-32P]ATP to the in vitro reaction mixture or 3′ end-labeling the phenol-extracted, ethanol-precipitated RNA with [32P]pCp and T4 RNA ligase (36). Both labeling procedures were followed by digestion with RNase A (cleaves RNA 3′ to pyrimidine residues) and RNase T1 (cleaves RNA 3′ to G residues) and resolution on 12% polyacrylamide sequencing gels containing 7 M urea (37).
RESULTS
PAP I Activity Is Associated with the Polysomes.
Polysomes isolated from SK5667 (PAP I+) rapidly incorporated [32P]AMP into a variety of RNA species (Fig. 1, lanes 4–7). In contrast, no significant incorporation was observed when the polysomes were prepared from SK8964 (PAP I−; Fig. 1, lanes 8–11). Incorporation was restored when SK8964 (PAP I−) was transformed with a six- to eight-copy number plasmid expressing PAP I (pJL89, pcnB+; Fig. 1, lanes 12–15). The addition of exogenous PAP I to wild-type polysomes further enhanced the incorporation of [32P]AMP (Fig. 1, lanes 1–3) as well as restoring polyadenylylating activity to PAP I− polysomes (data not shown). RNA species ranging in size from ≈80 nt to >2000 nt were radioactively labeled. Separation of the reaction mixtures on a 1.5% agarose gel suggested that the larger labeled species were mRNA and not the more abundant rRNAs (data not shown). The same distribution of [32P]AMP incorporation was seen when PAP I was produced by the chromosomal gene (Fig. 1, lanes 4–7), a plasmid-encoded gene, pJL89 (Fig. 1, lanes 12–15), or by the addition of exogenous PAP I to wild-type polysomes (Fig. 1, lanes 1–3).
Figure 1.
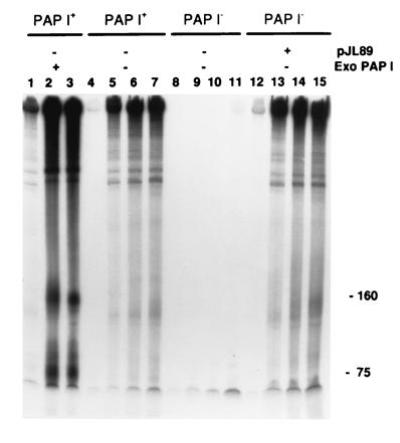
Incorporation of [32P]AMP into polysomal RNA. Polysomes were isolated from SK5667 (PAP I+) and SK8964 (PAP I−) and incubated in the presence of [α-32P]ATP. Samples were removed at the following time intervals. Lanes 1, 4, 8, and 12, 0.3 min; lanes 5 and 13, 2 min; lanes 2, 6, 9, and 14, 5 min; lanes 3, 7, 10, and 15, 10 min; and lane 11, 15 min. After phenol extraction and ethanol precipitation, the RNA was resolved by 6% PAGE in the presence of 7 M urea. Exogenous PAP I was added at the rate of 1 unit/reaction to lanes 1–3. Polysomes isolated from SK8964(ΔpcnB) transformed with pJL89, a plasmid carrying the pcnB+ gene, were used in lanes 12–15. RNA size markers in nucleotides are indicated on the right.
The salt requirements for optimal [32P]AMP incorporation (10 mM Mg2+/4 mM Mn2+/150 mM NaCl) and the reduced incorporation of either [32P]CMP or [32P]UMP (data not shown) agreed with the characteristics previously reported for partially purified PAP I (21–23, 38).
The association of PAP I activity exclusively with the polysomal material was demonstrated using a reconstitution experiment (Fig. 2). After a 10-min incubation of polysomes prepared from SK 8964 (PAP I−), both with and without an added S100 fraction from SK5667 (PAP I+), no significant [32P]AMP incorporation into higher molecular weight species was observed over a 20-fold range in protein concentration (Fig. 2, lanes 6–10). In the converse experiment, [32P]AMP was incorporated to the same extent by SK5667 (PAP I+) polysomes in the presence or absence of added S100 fractions from SK8964 (PAP I−; Fig. 2, lanes 1–5). This control demonstrated that the S100 from SK8964 (PAP I−) did not inhibit the activity of PAP I. Furthermore, no PAP I activity was detected in the S100 fraction derived from SK5667 (PAP I+) carrying pJL89 (pcnB+; data not shown).
Figure 2.
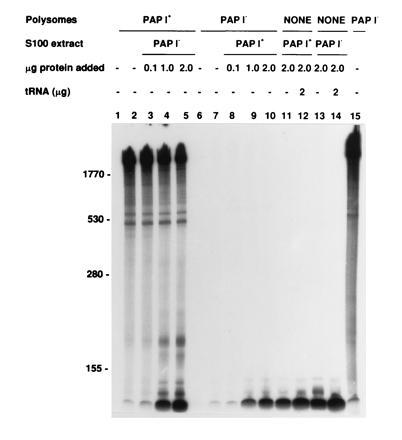
Incorporation of [32P]AMP into polysomal RNA in the presence or absence of postribosomal S100 supernatants. The experimental procedures are described in the text. Postribosomal extracts were made by centrifuging the lysed cells at 100,000 rpm for 2 hr. The supernatants (S100) were stored on ice overnight. PAP I+ is SK5667 (rna-19 thyA715) and PAP I− is SK8964 (ΔpcnB rna-19 thyA715). Lanes 1 and 6 were sampled at 0 min, and all remaining lanes were sampled at 10 min. Lanes 1–5, polysomes from PAP I+ to which the postribosomal supernatant from PAP I− was added at the indicated protein concentrations. Lanes 6–10, polysomes from PAP I− with postribosomal supernatant from PAP I+ added at the indicated protein concentrations. Lanes 11 and 12, postribosomal supernatant from PAP I+ without (lane 11) and with (lane 12) 2 μg of added tRNA. Lanes 13 and 14, postribosomal supernatant from PAP I− without (lane 13) and with (lane 14) 2 μg of added tRNA. Lane 15, polysomes from PAP I− incubated with 1 unit of exogenous PAP I enzyme. RNA size markers in nucleotides are shown on the left.
Although neither S100 extract incorporated [32P]-AMP into higher molecular weight RNA, some incorporation was visible at a size approximating tRNA (85 nt; Fig. 2, lanes 11–14). To determine if this incorporation resulted from the presence of nucleotidyl transferase (cca), we constructed SK8317 (Δcca ΔpcnB rna-19). The labeled low molecular weight species visible in Fig. 2 (lanes 3–14) were still observed using an S100 fraction from SK8317 (data not shown), indicating that some other enzyme activity was responsible for generating these species.
Decay of the lpp mRNA in Vitro.
The lpp gene encodes a major lipoprotein of the E. coli outer membrane and is transcribed into an abundant 322-nt transcript (39). It was detected on Northern blots of both total RNA and polysomal RNA from strain SK5667 (PAP I+) as a single band (Fig. 3 A–C), which decayed in vivo with a half-life of 10 ± 1.1 min at 37°C (Fig. 4A; Table 1). In SK8964 (PAP I−), the in vivo half-life of the full-length message increased to 14 ± 0.8 min (Fig. 4A; Table 1).
Figure 3.
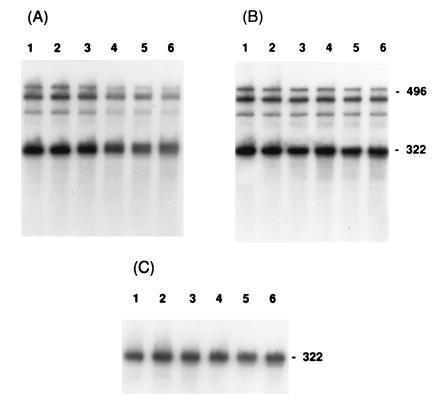
Northern blot analysis of lpp and trxA mRNA decay in vitro. Polysomes were isolated and incubated with or without ATP as described. Samples were removed at 0, 2, 4, 8 12, and 16 min (lanes 1–6, respectively), resolved using denaturing 6% PAGE, and probed with a full-length fragment for each respective gene. The top three bands represent trxA transcripts, the largest of which is 496 nt. lpp is the single 322-nt species. (A) Polysomes from SK5667 (PAP I+) incubated with ATP. (B) Polysomes from SK5667 (PAP I+) incubated without ATP. (C) Polysomes from SK8964 (PAP I−) incubated with ATP.
Figure 4.
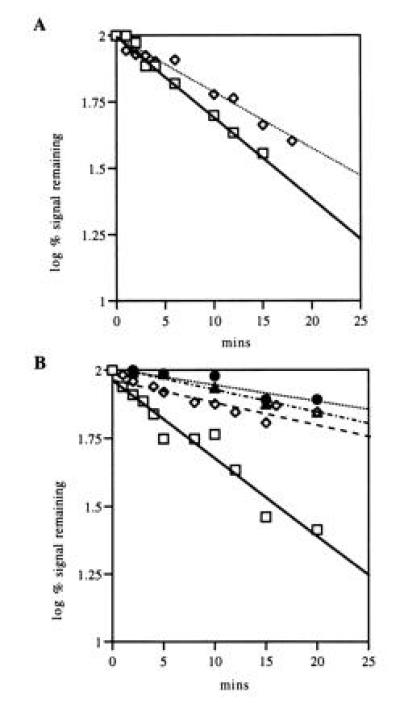
Decay of the lpp mRNA in vitro and in vivo. mRNA decay is plotted as the logarithm of the percent signal remaining versus time (2 = 100% while 1 = 10%). A linear regression analysis curve is drawn. (A) In vivo. Data was derived by analyzing Northern blots, such as those presented in Fig. 3, using a Molecular Dynamics model 400A PhosphoImager. □, SK5667 (PAP I+); and ◊, SK8964 (PAP I−). (B) In vitro. Polysomes were incubated with or without ATP. Samples were removed at the times indicated and analyzed using 6% denaturing PAGE. The Northern blots were analyzed as in A. □, Polysomes from SK5667 (PAP I+) plus ATP; ◊, polysomes from SK5667 (PAP I+) minus ATP; •, polysomes from SK8964 (PAP I−) plus ATP; and ▴, polysomes from SK8964 (PAP I−) minus ATP.
Table 1.
Half-lives of the trxA and lpp transcripts
| Gene | Time course, min | Strain
|
|||||
|---|---|---|---|---|---|---|---|
| SK5667
|
SK8964
|
||||||
| +ATP | −ATP | In vivo | +ATP | −ATP | In vivo | ||
| trxA | 5 | 6.4 ± 0.1 | 38 ± 12 | 2.6 ± 0.35 | 37 ± 13 | 33 ± 4.5 | 2.6 ± 0.1 |
| lpp | 20 | 10.3 ± 0.5 | 37 ± 6 | 10 ± 1.1 | 50 ± 11 | 37 ± 7 | 14 ± 0.8 |
Half-lives (in min) of the trx and lpp messages in vivo and in vitro, determined from the slope of the linear regression curve [y = mx − 1; slope = (log 0.5) × m]. Each half-life was independently determined a minimum of three times.
In the in vitro decay reaction, when polysomes from SK5667 (PAP I+) were incubated with ATP, the lpp transcript increased in size but decreased in intensity (Fig. 3A; the increase in size is more evident in Fig. 6). Decay of the message was linear (Fig. 4B) and gave a message half-life of 10.3 ± 0.3 min (Table 1). Decay was not observed to reach completion, with 10–20% of the signal always remaining. When ATP was not included in the reaction mixture, no increase in size of the message was seen (Fig. 3B), and decay was very slow with a half-life of longer than 37 ± 6 min (Fig. 4B; Table 1). No decay products were detected in any of the reactions (Fig. 3). Incubation of polysomes from SK8964 (PAP I−) both with (Fig. 3C) and without ATP resulted in very slow decay of the full-length message, giving half-lives of >37 min (Fig. 4B; Table 1). Message decay was restored by transformation of SK8964 with pJL89 (pcnB+; data not shown).
Figure 6.
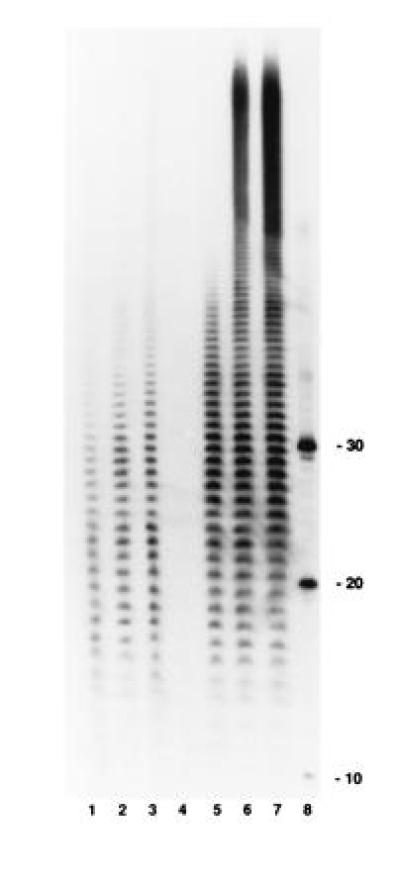
Poly(A) tails formed during in vitro mRNA decay. Polysomes were incubated in the presence of [α-32P]ATP. Samples were removed at various times, deproteinated, and then digested exhaustively with RNase A and RNase T1 as described. An autoradiogram of a 12% polyacrylamide gel is shown. Lanes 1–3, SK5667 (PAP I+) at 2, 5, and 10 min, respectively; lane 4, SK8964 (PAP I−) at 10 min; lanes 5–7, SK8964 (PAP I−) with 0.4 units of exogenous PAP I per reaction at 2, 5, and 10 min, respectively; and lane 8, 10-, 20-, and 30-mers of oligo(dA) end-labeled with [γ-32P]ATP.
Decay of the trxA Message in Vitro.
To determine whether the observations described above were limited to the lpp transcript, we examined the decay of a second message. Transcription of the thioredoxin gene (trxA), encoding a small nonessential protein, yields three distinct bands on Northern blots, the largest of which is 496 nt (7). In SK5667 (PAP I+), the in vivo decay at 37°C of this message was linear and rapid, with a half-life of 2.6 ± 0.35 min (Table 1). No decay intermediates were visible (data not shown). In SK8964 (PAP I−), the half-life of the full-length trxA message was also 2.6 ± 0.1 min at 37°C (Table 1). Polysomes isolated from these two strains showed the same trxA transcript pattern as observed in vivo (Fig. 3A).
In the in vitro decay reaction, when polysomes isolated from SK5667 (PAP I+) were incubated in the presence of ATP, decay was again observed (Fig. 3A, +ATP; Fig. 3B, −ATP). The decay curve for the trxA message was linear for the first 5 min, but with 50% of the message remaining, the rate of decay slowed, approaching that observed without ATP (data not shown). The rate of decay of the trxA message was calculated for the first 5 min and was 6.4 ± 0.1 min with ATP and 38 ± 12 min without ATP (Table 1). Polysomes isolated from SK8964 (PAP I−) gave trxA half-lives of 37 ± 13 and 33 ± 4.5 min, with and without ATP, respectively (Table 1). In a control experiment, polysomes from SK5667 were mixed with either an S30 or S100 extract from SK5667 in the absence of ATP. No appreciable trxA decay was observed over a 60-min incubation (data not shown).
In Vitro Polyadenylylation Is Specific to mRNAs.
Although [32P]AMP was incorporated into high molecular weight species, the data presented in Fig. 1 does not distinguish between the polyadenylylation of mRNA and ribosomal RNA. Accordingly, we directly examined the 3′ end of 23S rRNA, as well as the lpp and trxA mRNAs for the presence of poly(A) tails. We compared the electrophoretic mobility of the various specific RNAs before and after treatment with oligo (dT)20 and RNase H. Because the lpp transcript is only 322 nt long, alterations in its electrophoretic mobility could be visualized directly. In the case of 23S rRNA, an oligonucleotide complementary to nucleotides 6193–6212 (40) was used in the presence of RNase H to generate a 189-nt oligonucleotide containing the mature 3′ end.
The results of one such experiment, in which duplicate samples were probed for either lpp or 23S rRNA, are shown in Fig. 5. Using SK5667 (PAP I+) polysomes, the 322-nt lpp transcript clearly increased in size after the 10-min incubation with ATP (Fig. 5A, lane 2). After digestion with RNase H and oligo(dT)20, the RNA returned to the size obtained from the 0-min control (Fig. 5A, lane 3). No increase in size was observed with polysomes prepared from SK8964 (PAP I−; Fig. 5A, lanes 4–6). When exogenous PAP I was added to SK8964 polysomes (Fig. 5A, lanes 7–9), the size of the lpp mRNA increased in a manner similar to that observed with wild-type polysomes (Fig. 5A, compare lane 2 with lane 8). RNase H digestion in the presence of oligo(dT)20 shortened the RNA back to its original length (Fig. 5A, lane 9). Comparable results were obtained for the trxA mRNA (data not shown).
Figure 5.
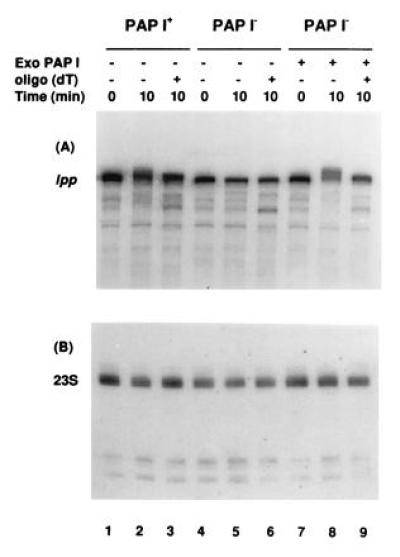
Polyadenylylation in polysomes occurs on mRNA and not rRNA. RNA was isolated and treated as described. Following electrophoresis on 6% denaturing PAGE, the Northern blots were probed with a full-length lpp probe (A) or an oligonucleotide complementary to the 3′ end of the mature 23S rRNA (B). Exogenous PAP was added as indicated at 0.4 units/reaction. All samples were digested with RNase H.
In contrast, the 189-nt 3′ terminal fragment of 23S rRNA obtained with specific RNase H digestion showed no evidence of any increase in size either after incubation of SK5667 (PAP I+) polysomes with ATP (Fig. 5B, lanes 1–3) or in the presence of exogenously added PAP I, (Fig. 5B, lanes 7–9). A poly(A) tail as short as 10 nt would have caused a detectable electrophoretic shift (data not shown).
Analysis of Poly(A) Tails Generated in Vitro.
The addition of poly(A) tails to RNA during the incubation of polysomes from SK5667 (PAP I+) was confirmed using either [α-32P]ATP or [32P]pCp and T4 RNA ligase 3′-end labeling as described in Materials and Methods. RNA was extracted from samples treated as described in Fig. 1 (lanes 4–7) and subjected to exhaustive digestion with RNase A and RNase T1. As shown in Fig. 6, in SK5667 (PAP I+) the amount and length of the poly(A) tails increased with time. The maximum length was ≈50 nt (Fig. 6, lanes 2 and 3). In contrast, in SK8964 (PAP I−), no poly(A) tails were detected after a 10-min incubation (Fig. 6, lane 4), but extensive polyadenylylation occurred either when 0.4 units of exogenous PAP I was added to each sample (Fig. 6, lanes 5–7) or when polysomes were prepared from SK8964 transformed with pJL89 (PAP I+).
The time course of polyadenylylation was also followed using [32P]pCp end-labeling. This technique permitted us to determine the level of endogenous poly(A) tails before the addition of ATP. As shown in Fig. 7 (lanes 2–5), the polyadenylylation profile was comparable to that observed using direct labeling (Fig. 6, lanes 1–3). Of particular interest was the observation that the bulk of endogenous poly(A) tails were not associated with the polysomes but with the postribosomal supernatant (Fig. 7, compare lanes 2 and 9). In addition, exogenous PAP I caused a massive increase in the extent and size of the poly(A) tails (Fig. 7, lanes 6–8).
Figure 7.
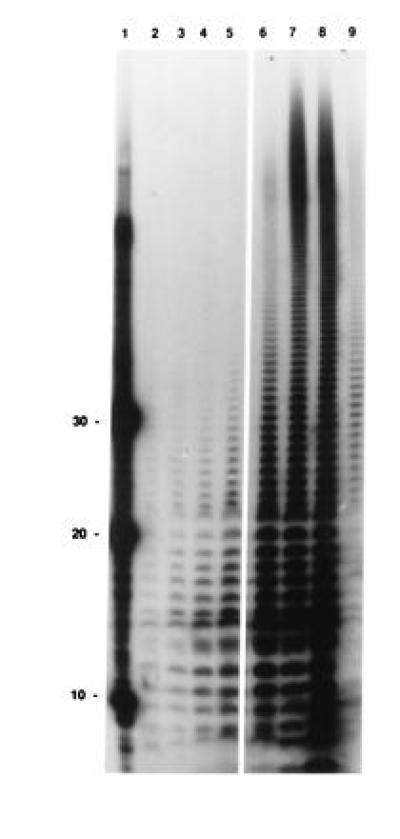
Measurement of endogenous poly(A) tails in polysomal RNA. Samples were taken from an in vitro reaction in which polysomes were incubated in the presence of ATP. Poly(A) tails were visualized by [32P]pCp end-labeling in the presence of T4 RNA ligase, followed by digestion with RNase A and RNase T and resolution by 12% PAGE, as described. Lane 1, 10-, 20-, and 30-mer end-labeled oligomers of oligo(dA); lanes 2–5, SK5667 (PAP I+) sampled at 0, 2, 5, and 10 min, respectively; lanes 6–8; SK8964 (PAP I−) plus 0.4 units of exogenous PAP at 2, 5, and 10 min, respectively; and lane 9, postribosomal supernatant from SK5667 (PAP I+). Lanes 2 and 9 contain 20 μg of RNA, all others lanes have 10 μg of RNA.
In Vitro mRNA Decay Rates Are Not Affected by Exogenously Added PAP I.
To determine if the level of PAP I was rate-limiting in the in vitro reaction, we added different concentrations of exogenous PAP I to polysomes from either SK5667 (PAP I+) or SK8964 (PAP I−). We measured total polyadenylylation, as well as determining the half-lives of both the lpp and trxA transcripts. While the presence of 0.4 units of exogenous PAP I led to a large increase in polyadenylylation in both PAP I− (Fig. 7, lanes 7–9) and PAP I+ (data not shown) polysomes, the decay rates for the lpp and trxA mRNAs were not significantly changed relative to those observed in the absence of enzyme. Thus SK5667 (PAP I+) polysomes in the presence of 0.4 units of PAP I yielded a half-life comparable to that reported in Table 1. Interestingly, with SK8964 (PAP I−) polysomes, mRNA decay was not restored by the exogenous PAP I, even though the lpp and trxA transcripts were extensively polyadenylylated (data not shown).
DISCUSSION
By employing polysomes as the source of both mRNAs and the enzymes necessary to modify and degrade them, we have been able to develop an in vitro decay system in which the lpp and trxA transcripts are degraded at relative rates comparable to those determined in vivo (Fig. 4; Table 1). By using either wild-type (SK5667, PAP I+) or ΔpcnB (SK8964, PAP I−) strains of E. coli as the source of the polysomes, we have demonstrated that mRNA decay can be triggered by the formation of poly(A) tails. This observation confirms the reports that poly(A) tails are important for mRNA decay in vivo (17, 18).
The poly(A) tails formed in the in vitro reactions are up to 50 nt long (Figs. 6 and 7), which is in good agreement with the sizes previously reported in vivo (17, 18). Although poly(A) tails were easily detected on the lpp (Fig. 5A) and trxA transcripts (data not shown), no tailing was detected on the mature 3′ end of 23S RNA (Fig. 5B). These results suggest that in vitro poly(A) tails are specifically added to mRNAs, triggering their decay. The requirement for polyadenylylation before significant decay can be detected in vitro can be explained by the fact that the bulk of the poly(A) tails observed in vivo are found in the postribosomal supernatant and are not associated with the polysomes (Fig. 7, lanes 2 and 9).
The decay rate of the lpp mRNA in the in vitro system was almost identical with the rate determined in vivo (Table 1), and between 80–90% of the transcript was degraded in the reaction. These observations suggest that poly(A)-directed decay may be the primary pathway for degradation of this mRNA and that in vitro this system is operating effectively. This hypothesis is supported by the in vivo data in which the absence of PAP I results in a reduced rate of mRNA decay (Fig. 4A; Table 1; ref. 17).
On the other hand, the results obtained with trxA show a 2.4-fold difference in the in vivo (2.6 min) versus the in vitro (6.4 min) half-life. In addition, the decay of the message was more limited in extent (≈50%). This may mean that, for trxA, poly(A)-directed decay is not the primary pathway in vivo. This idea is supported by the fact that there is no change in the in vivo half-life in the absence of PAP I (Table 1). Furthermore, O’Hara et al. (17) showed less of an effect of PAP I on trxA mRNA decay in multiple mutants compared with the lpp and ompA transcripts in vivo. Thus, with the shorter-lived trxA transcript, a poly(A)-independent decay system may function equally well in the absence of PAP I. Taken together, the data from lpp and trxA suggest the existence of alternative pathways that are efficient for some messages but not for others.
The disappearance of full-length mRNAs, with no significant appearance of decay intermediates (Fig. 3A), suggests that the rest of the poly(A)-directed decay system is also associated with the polysomes. Endonucleolytic cleavages cannot be excluded, but the absence of distinct intermediates strongly indicates that exonucleolytic degradation rapidly destroys the transcripts. However, since the in vitro system has been optimized for poly(A) tail formation, it is probable that some components of the poly(A) directed decay system are either limited in activity or missing.
In particular, the absence of phosphate in the reaction mixture would be expected to inhibit the phosphorolytic activity of polynucleotide phosphorylase (PNPase; ref. 1). This is significant because there is in vivo evidence that PNPase is the primary enzyme responsible for poly(A) tail degradation (ref. 17; J. Chekanova, B. Mohanty, and S.R.K., unpublished results). Furthermore, it is not known whether inhibition of the phosphorolytic activity of PNPase might affect the formation of the RNase E/PNPase multiprotein complex (41–44). Such limitations could account for the steady in vitro accumulation of poly(A) tails with time (Fig. 3 A and B, lanes 5 and 6; Fig. 5).
Although we initially assumed that the addition of the poly(A) tail might be the rate-limiting step in decay, the results presented here demonstrate a more complex relationship between polyadenylylation and mRNA decay. Specifically, even though we could dramatically increase polyadenylylation levels by adding exogenous PAP I to either PAP I+ (data not shown) or PAP I− polysomes (Fig. 7, lanes 7–9), mRNA decay rates did not increase relative to those observed in the absence of added enzyme (data not shown). These observations suggest that the presence or absence of the ribonucleases or associated factors responsible for mRNA decay triggered by the formation of a poly(A) tail require the concomitant association of PAP I. Thus polyadenylylation of mRNA may be a prerequisite for decay but is not sufficient in itself. An alternative explanation, although not as attractive, is that the ATP is utilized by a different decay factor whose association with polysomes is dependent on the presence of PAP I.
Another issue to consider is the quantitative association of PAP I with the polysomal fraction (Figs. 1 and 2), even when the cell is overproducing the enzyme (data not shown). Throughout this work we have assumed that PAP I is directly associated in some fashion with 70S ribosomes, in part because we used the accepted method employed to isolate polysomes. Although previously reported (22), this association was considered to be fortuitous. There is precedent for such apparent random associations since the periplasmic RNase I is found associated with the polysomal fraction in E. coli crude extracts (45). Since a role in mRNA decay makes such an association very significant, it will be necessary to obtain direct genetic evidence to confirm the association of PAP I with polysomes rather than a nonspecific interaction.
As mentioned above, our current results cannot distinguish between decay as a result of a combination of endo- and exonucleolytic cleavages or rapid exonucleolytic decay. The availability of various multiple mutants in which PNPase, RNase II, RNase E, and RNase III can be inactivated should help answer this question. More importantly, the system may help to identify additional ribonucleases involved in mRNA decay. Babitzke et al. (46) demonstrated that mRNA decay still occurred relatively efficiently in a PNPase, RNase II, RNase E, RNase III quadruple mutant. This observation clearly demonstrates that there are additional enzymes involved in mRNA decay.
It should also be noted that this system is not currently constituted to permit active translation in vitro. Although conflicting data have been reported regarding the importance of translation relative to mRNA decay (20), it is clear that translation does have an effect (16). In fact, the absence of translation may be the reason that the lpp and trxA mRNAs do not decay to completion. The static ribosomes may prevent access for endo- and exonucleases regardless of the existence of a poly(A) tail. Furthermore, it will make it possible to test the hypothesis that translation may actually be a requirement for polyadenylylation. Taken together, the in vitro system described above provides the basis of a powerful approach for analyzing the biochemical features of mRNA decay in E. coli.
Acknowledgments
The authors thank Kaija Lewis and Ze’eva Kushner for technical assistance and Eileen O’Hara for invaluable advice and support. This work was supported by a National Institutes of Health grant to S.R.K. (GM28760).
Footnotes
Abbreviations: PAP, poly(A) polymerase; PNPase, polynucleotide phosphorylase.
References
- 1.Grunberg-Manago M. Prog Nucleic Acids Res. 1963;1:93–133. [Google Scholar]
- 2.Nikolaev N, Folsom V, Schlessinger D. Biochem Biophys Res Commun. 1976;70:920–924. doi: 10.1016/0006-291x(76)90679-3. [DOI] [PubMed] [Google Scholar]
- 3.Reiner A M. J Bacteriol. 1969;97:1437–1443. doi: 10.1128/jb.97.3.1437-1443.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghora B K, Apirion D. Cell. 1978;15:1055–1066. doi: 10.1016/0092-8674(78)90289-1. [DOI] [PubMed] [Google Scholar]
- 5.Kuwano M, Ono M, Endo H, Hori K, Nakamura K, Hirota Y, Ohnishi Y. Mol Gen Genet. 1977;154:279–285. doi: 10.1007/BF00571283. [DOI] [PubMed] [Google Scholar]
- 6.Donovan W P, Kushner S R. Proc Natl Acad Sci USA. 1986;83:120–124. doi: 10.1073/pnas.83.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arraiano C M, Yancey S D, Kushner S R. J Bacteriol. 1988;170:4625–4633. doi: 10.1128/jb.170.10.4625-4633.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mudd E A, Krisch H M, Higgins C F. Mol Microbiol. 1990;4:2127–2135. doi: 10.1111/j.1365-2958.1990.tb00574.x. [DOI] [PubMed] [Google Scholar]
- 9.Meyer B J, Schottel J L. Mol Microbiol. 1992;6:1095–1104. doi: 10.1111/j.1365-2958.1992.tb01547.x. [DOI] [PubMed] [Google Scholar]
- 10.Arraiano C M, Yancey S D, Kushner S R. J Bacteriol. 1993;175:1043–1052. doi: 10.1128/jb.175.4.1043-1052.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melefors Ö, von Gabain A. Cell. 1988;52:893–901. doi: 10.1016/0092-8674(88)90431-x. [DOI] [PubMed] [Google Scholar]
- 12.Belasco J G, Nilsson G, von Gabain A, Cohen S N. Cell. 1986;46:245–251. doi: 10.1016/0092-8674(86)90741-5. [DOI] [PubMed] [Google Scholar]
- 13.Newbury S F, Smith N H, Robinson E C, Hiles I D, Higgins C F. Cell. 1987;48:297–310. doi: 10.1016/0092-8674(87)90433-8. [DOI] [PubMed] [Google Scholar]
- 14.Emory S A, Belasco J G. J Bacteriol. 1990;172:4472–4481. doi: 10.1128/jb.172.8.4472-4481.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plamann M D, Stauffer G V. Mol Gen Genet. 1990;220:301–306. doi: 10.1007/BF00260498. [DOI] [PubMed] [Google Scholar]
- 16.Iost I, Dreyfus M. EMBO J. 1995;14:3252–3261. doi: 10.1002/j.1460-2075.1995.tb07328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Hara E B, Chekanova J A, Ingle C A, Kushner Z R, Peters E, Kushner S R. Proc Natl Acad Sci USA. 1995;92:1807–1811. doi: 10.1073/pnas.92.6.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Häjnsdorf E, Braun F, Haugel-Nielsen J, Regnier P. Proc Natl Acad Sci USA. 1995;92:3973–3977. doi: 10.1073/pnas.92.9.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ross J, Kobs G, Brewer G, Peltz S W. J Biol Chem. 1987;262:9374–9381. [PubMed] [Google Scholar]
- 20.Peterson C. In: Control of Messenger RNA Stability. Belasco J, Brawerman G, editors. San Diego: Academic; 1993. pp. 117–148. [Google Scholar]
- 21.August J, Ortiz P J, Hurwitz J. J Biol Chem. 1962;237:3786–3793. [PubMed] [Google Scholar]
- 22.Hardy S J S, Kurland C G. Biochemistry. 1966;11:3676–3684. doi: 10.1021/bi00875a042. [DOI] [PubMed] [Google Scholar]
- 23.Cao G-J, Sarkar N. Proc Natl Acad Sci USA. 1992;89:10380–10384. doi: 10.1073/pnas.89.21.10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu F, Lin-Chao S, Cohen S N. Proc Natl Acad Sci USA. 1993;90:6756–6760. doi: 10.1073/pnas.90.14.6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu F, Cohen S N. Nature (London) 1995;374:180–183. doi: 10.1038/374180a0. [DOI] [PubMed] [Google Scholar]
- 26.Kushner S R. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. 2nd Ed. Neidhardt F C, Curtiss R III, Ingraham J L, Lin E C C, Low J, K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 849–860. [Google Scholar]
- 27.Gesteland R F. J Mol Biol. 1966;16:67–84. doi: 10.1016/s0022-2836(66)80263-2. [DOI] [PubMed] [Google Scholar]
- 28.Willetts N S, Mount D W. J Bacteriol. 1969;100:923–934. doi: 10.1128/jb.100.2.923-934.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wright M. J Bacteriol. 1971;107:87–94. doi: 10.1128/jb.107.1.87-94.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Parkinson J S. J Bacteriol. 1989;171:1254–1261. doi: 10.1128/jb.171.3.1254-1261.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ron E Z, Kohler R E, Davis B D. Science. 1966;153:1119–1120. doi: 10.1126/science.153.3740.1119. [DOI] [PubMed] [Google Scholar]
- 32.Miller J H. Experiments in Molecular Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1972. [Google Scholar]
- 33.Chapman J M, Ayrey G. The Use of Radioactive Isotopes in the Life Sciences. London: Allen & Unwin; 1982. [Google Scholar]
- 34.Donis-Keller H. Nucleic Acids Res. 1979;7:179–192. doi: 10.1093/nar/7.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Narayan P, Ayers D F, Rottmann F M, Maroney P A, Nilsen T W. Mol Cell Biol. 1987;7:1572–1575. doi: 10.1128/mcb.7.4.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahlquist P, Kaesburg P. Nucleic Acids Res. 1979;7:1195–1204. doi: 10.1093/nar/7.5.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sachs A B, Davis R W. Cell. 1989;58:857–868. doi: 10.1016/0092-8674(89)90938-0. [DOI] [PubMed] [Google Scholar]
- 38.Ramanarayanan M, Srinivasan P R. J Biol Chem. 1976;251:6274–6286. [PubMed] [Google Scholar]
- 39.Hirashima A, Childs G, Inouye M. J Mol Biol. 1973;79:373–389. doi: 10.1016/0022-2836(73)90012-0. [DOI] [PubMed] [Google Scholar]
- 40.Brosius J, Dull T J, Sleeter D D, Noller H F. J Mol Biol. 1981;148:107–127. doi: 10.1016/0022-2836(81)90508-8. [DOI] [PubMed] [Google Scholar]
- 41.Py B, Causton H, Mudd E A, Higgins C F. Mol Microbiol. 1994;14:717–729. doi: 10.1111/j.1365-2958.1994.tb01309.x. [DOI] [PubMed] [Google Scholar]
- 42.Carpousis A J, Van Houwe G, Ehretsmann C, Krisch H M. Cell. 1994;76:889–900. doi: 10.1016/0092-8674(94)90363-8. [DOI] [PubMed] [Google Scholar]
- 43.Py B, Higgins C F, Krisch H M, Carpousis A J. Nature (London) 1996;381:169–172. doi: 10.1038/381169a0. [DOI] [PubMed] [Google Scholar]
- 44.Miczak A, Kaberdin V R, Wei C-L, Lin-Chao S. Proc Natl Acad Sci USA. 1996;93:3865–3869. doi: 10.1073/pnas.93.9.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neu H C, Heppel L A. Biochemistry. 1964;51:1267–1274. doi: 10.1073/pnas.51.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Babitzke P, Granger L, Kushner S R. J Bacteriol. 1993;175:229–239. doi: 10.1128/jb.175.1.229-239.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]