

Abstract
A protein fluorescence probe system, coupling excited-state intermolecular Förster energy transfer and intramolecular proton transfer (PT), is presented. As an energy donor for this system, we used tryptophan, which transfers its excitation energy to 3-hydroxyflavone (3-HF) as a flavonol prototype, an acceptor exhibiting excited-state intramolecular PT. We demonstrate such a coupling in human serum albumin–3-HF complexes, excited via the single intrinsic tryptophan (Trp-214). Besides the PT tautomer fluorescence (λmax = 526 nm), these protein–probe complexes exhibit a 3-HF anion emission (λmax = 500 nm). Analysis of spectroscopic data leads to the conclusion that two binding sites are involved in the human serum albumin–3-HF interaction. The 3-HF molecule bound in the higher affinity binding site, located in the IIIA subdomain, has the association constant (k1) of 7.2 × 105 M−1 and predominantly exists as an anion. The lower affinity site (k2 = 2.5 × 105 M−1), situated in the IIA subdomain, is occupied by the neutral form of 3-HF (normal tautomer). Since Trp-214 is situated in the immediate vicinity of the 3-HF normal tautomer bound in the IIA subdomain, the intermolecular energy transfer for this donor/acceptor pair has a 100% efficiency and is followed by the PT tautomer fluorescence. Intermolecular energy transfer from the Trp-214 to the 3-HF anion bound in the IIIA subdomain is less efficient and has the rate of 1.61 × 108 s−1, thus giving for the donor/acceptor distance a value of 25.5 Å.
Excitation by light may initiate both intramolecular and intermolecular transformations of a molecule. Among excited-state intramolecular processes, the most important are electron transfer (1–4), proton transfer (PT; refs. 5 and 6), and conformational transformation (e.g., twisting, torsion, and isomerization; refs. 7–9). The most conspicuous intermolecular events are electron, proton, and energy transfer (10, 11). Excited-state transformations can be coupled with each other and with the surrounding medium. Dependence of the excited-state intramolecular electron transfer on environmental relaxation (2, 3) and the twisting of solute groups in response to the excited-state charge transfer (12–15) are well-established phenomena. When joined in one molecular system, excited-state events can compete dynamically. An example of such competition is the mutual exclusion of intramolecular electron and PT in 4′-diethylamino-3-hydroxyflavone (16–19). Proper pairing of excited-state transformations can be a source of structural/dynamical information about the molecules possessing these transformations, the excited-state transformations themselves, and the medium where the coupling takes place.
The present paper focuses on the coupling between the Förster intermolecular energy transfer (11, 20, 21) and intramolecular PT (refs. 6 and 22; Fig. 1). The first step of such a coupling is the R−6-dependent (R is the intermolecular distance) dipole–dipole intermolecular energy transfer from excited donor to unexcited acceptor. Förster energy transfer occurs for the very weak range of dipole–dipole interaction energies (10−1–101 cm−1) and has a rate range of 106–1011 s−1 (20). Generation of the acceptor excited state (S1) may initiate in the latter an intramolecular excited-state PT with the final formation of the PT-excited tautomer (S1′). PT-excited tautomer may emit light undergoing transition to the PT tautomer ground state (S0′), followed by a reverse PT to the normal tautomer ground state (S0). Excited-state PT occurs in femtoseconds to picoseconds (23, 24) and results in a PT tautomer fluorescence that is shifted from the normal tautomer absorption by 8000–13,000 cm−1.
Figure 1.
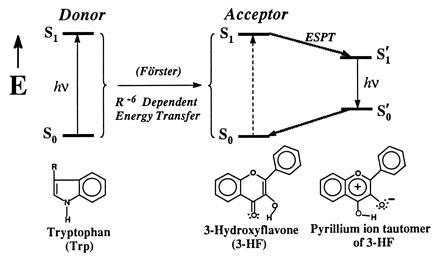
Coupling between intermolecular Förster-type energy transfer and intramolecular PT. Donor transfers UV energy with an R−6 dependence to acceptor, undergoing intramolecular PT and emitting in the visible region.
As a donor for the system coupling intermolecular energy transfer and intramolecular PT, we chose tryptophan. This molecule efficiently absorbs and emits light in the UV spectral region. A molecule capable of exhibiting intramolecular excited-state PT, which absorbs light in the same region where tryptophan emits (the critical Förster criterion), is 3-hydroxyflavone (3-HF; ref. 22). We chose 3-HF both as a PT fluorescence probe and also as a prototype for biologically significant flavonols. Coupling between intermolecular energy transfer and excited-state PT may transform the excitation energy of tryptophan (λex = 280–290 nm) to the green fluorescence of the PT tautomer of 3-HF over distances appropriate for efficient Förster energy transfer (≈50 Å). As an environment for the coupling between intermolecular energy transfer and intramolecular PT, we used a protein, human serum albumin (HSA). This protein easily binds various agents and transports them in the blood stream. HSA contains a single intrinsic tryptophan and has internal dimensions (25) favorable for the Förster type of energy transfer.
MATERIALS AND METHODS
Spectroscopic Measurements.
The absorption spectra were recorded on a Shimadzu UV-2100 Spectrophotometer. The fluorescence spectra and time-resolved phase-modulation experiments were performed with a Fluorolog-2 Spectrofluorometer (Spex Industries, Edison, NJ).
Preparation of HSA–3-HF Complexes.
The commercial product of 3-HF (Aldrich) was recrystallized twice from methanol and once from cyclohexane and then vacuum-sublimed. Essentially fatty acid-free HSA was a product from Sigma. The 3-HF was dissolved in ethanol, and then 3 μl of the stock solution was added to the protein solution in 0.05 M Hepes buffer (pH 7.0; 2.2 ml). After incubation and mixing for 3 h at room temperature, the solutions were centrifuged for 30 min at 23,000 × g.
Distance Determination for the HSA–3-HF Complexes.
Distance between the donor/acceptor pair for the HSA–3-HF complexes was determined utilizing the Förster theory of excitation energy transfer (11). According to this theory, the rate of energy transfer from an excited donor to an unexcited acceptor is given by the following equation:
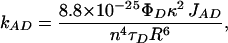 |
1 |
where τD is the donor fluorescence lifetime, ΦD is the donor quantum yield, n is the refractive index of the medium between the donor and acceptor, κ is the dipole–dipole orientation factor, R is the distance between the donor and acceptor, and JAD is the overlap integral between the donor fluorescence spectrum and acceptor absorption spectrum:
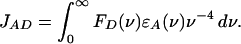 |
2 |
The distance R0 at which the rate of energy transfer is equal to the sum of the rates of all other deexcitation modes of the donor is called the Förster radius:
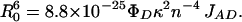 |
3 |
RESULTS AND DISCUSSION
Stoichiometry of HSA–3-HF Interaction.
HSA has two principal binding sites located in the IIA and IIIA subdomains (25). The single intrinsic tryptophan of this protein, Trp-214, is situated in the IIA subdomain, where Lys-199 and His-242 play a key role in the protein–ligand interaction. The most important residues of the IIIA subdomain binding site are Tyr-411 and Arg-410. Fig. 2 shows the absorption spectra of HSA–3-HF complexes formed at 1:1 and 1:2 protein–probe concentration ratios. In both cases, the absorption spectra consist of three distinct bands. Comparison of these bands with the 3-HF absorption in aprotic and protic solvents (26–28) suggests that both 3-HF normal tautomer (λmax = 345 nm) and anion (λmax = 417 nm) contribute to the probe absorption. Large spectral shift between the ionic forms of 3-HF allows to separate their relative contributions to the absorption of HSA–3-HF complexes (Fig. 2) and determine concentrations of both forms in the protein–probe solutions (see A). Employing absorption of individual species of 3-HF and assuming that each binding site of HSA hosts predominantly one of the two ionic forms of the probe, depending on specific interactions of the ligand with surrounding amino acid residues, we determined the association constants for each of the two binding sites involved (see A). The higher affinity site of HSA has association constant (k1) of 7.2 × 105 M−1 and predominantly hosts the 3-HF anion, while the lower affinity site (k2 = 2.5 × 105 M−1) is occupied by the normal tautomer. Since the aforementioned constants have the same order of magnitude, the HSA–3-HF complexes formed at the 1:1 protein–ligand concentration ratio contain 3-HF molecules bound in the higher affinity as well as lower affinity binding sites. Fluorescence spectra of HSA–3-HF complexes exhibit dependence on excitation wavelength characteristic of a two component mixture with dominant contributions coming from the PT tautomer and the anion (Fig. 3). Excitation at 350 nm corresponding to the absorption maximum of the 3-HF normal tautomer and minimum of the anion yields a fluorescence of almost pure PT tautomer (λmax = 526 nm). Excitation at the anion absorption maximum, 420 nm, reveals fluorescence of pure anionic form (λmax = 500 nm).
Figure 2.
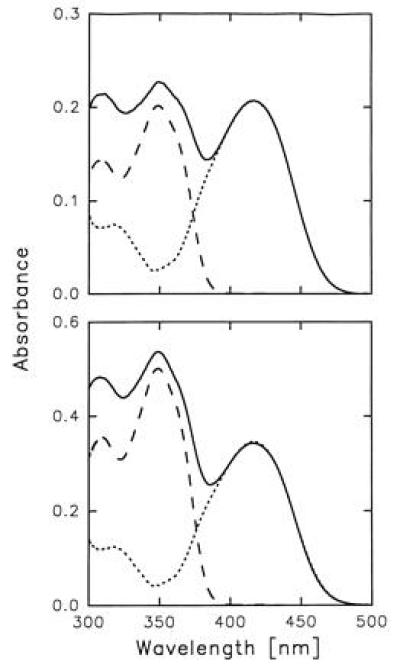
Absorption spectra of HSA–3-HF complexes at 298 K. (Upper) [HSA] = 40 μM, [3-HF] = 40 μM; (Lower) [HSA] = 40 μM, [3-HF] = 80 μM. Decomposition of the absorption spectra into the two components corresponding to the two ionic forms of 3-HF is shown by --- for 3-HF normal tautomer; and ··· for 3-HF anion.
Figure 3.
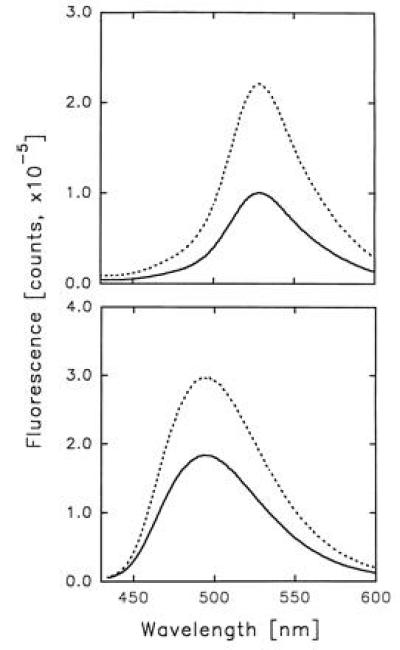
Fluorescence of HSA–3-HF complexes excited at 350 nm (Upper) and 420 nm (Lower). ——, [HSA] = [3-HF] = 40 μM; ···, [HSA] = 40 μM, [3-HF] = 80 μM.
Excited-State Intermolecular Energy Transfer in HSA–3-HF Complexes.
3-HF molecules bound to HSA cause an efficient quenching of the tryptophan fluorescence (Fig. 4). Simultaneously, the probe emission excited via the intermolecular energy transfer from Trp-214 appears. The latter can be decomposed into the two components arriving from the 3-HF PT tautomer and anion (Fig. 5). Relative contributions of the two ionic forms to the 3-HF fluorescence are determined by the occupancy of the two principle binding sites of HSA with the probe molecules and by the rates of energy transfer from Trp-214 to the bound ligands. As Fig. 5 shows, the PT fluorescence dominates over the anion emission for the HSA–3-HF complexes formed at 1:1 and 1:2 protein–probe concentration ratio and excited at 297 nm, indicating much faster energy transfer from Trp-214 to a normal tautomer than to an anion. That suggests that the site hosting a normal tautomer is much closer to Trp-214 than the one binding an anion. It appears reasonable to associate the 3-HF normal tautomer binding site with the tryptophan-containing IIA subdomain characterized by x-ray crystallography (25) and assume that the bound ligand is located in close proximity from Trp-214, which enables very fast and efficient energy transfer. On the other hand, the energy transfer to 3-HF anion bound in the higher affinity site has much lower rate though it is still fast enough to compete with the Trp-214 fluorescence in complexes with a vacant lower affinity site. The data on HSA fluorescence quenching allows the determination of the rate of energy transfer from Trp-214 to the 3-HF anion, ketA (see B). The ketA value of 1.61 × 108 s−1 can be used to estimate the distance between Trp-214 and 3-HF anion situated in the IIIA subdomain. Employing a water solution of tryptophan as reference [Φ = 0.14 (29)], we determined the fluorescence quantum yield of HSA tryptophan to be 0.11. The modeling of HSA fluorescence decay data (λex = 297 nm), obtained in the phase-modulation experiments, yields the best fit for the double-exponential decay with the following parameters: α1 = 0.8, τ1 = 6.2 ns; α2 = 0.2, τ2 = 1.5 ns; χ2 = 0.6. For the distance determination, we used the average integral lifetime of HSA tryptophan of 4.5 ns, giving the best fit for the one-exponential decay model. A refractive index value equal to that of water (n = 1.4) and an orientation factor κ2 = 2/3 were employed. The value of Förster radius for the Trp-214–3-HF anion donor/acceptor pair was determined to be 25.4 Å, indicating the 25.5 Å separation between Trp-214 and 3-HF anion (see B).
Figure 4.
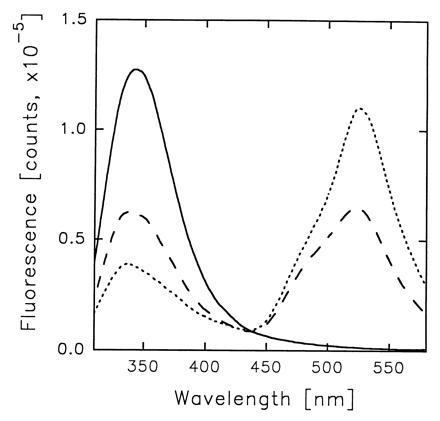
Fluorescence spectra of HSA (40 μM) in absence (——) and presence of 40 μM (---) and 80 μM (···) 3-HF. λex = 297 nm.
Figure 5.
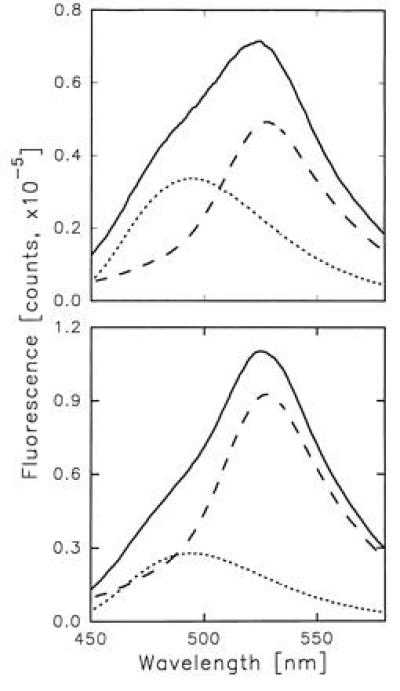
Component analysis of 3-HF fluorescence excited via the energy transfer from Trp-214: (Upper) [HSA] = [3-HF] = 40 μM; (Lower) [HSA] = 40 μM, [3-HF] = 80 μM. ——, Total fluorescence of 3-HF; ---, PT tautomer of 3-HF; and ···, 3-HF anion. λex = 297 nm.
Summing up the results on the HSA–3-HF complexes, we propose the following model. The higher affinity binding site with association constant of 7.2 × 105 M−1 is located in the IIIA subdomain. At this site, the 3-HF molecule is bound predominantly in anionic form. Tyr-411 is the most probable candidate for the 3-HF anion formation in this site, since the phenolic hydroxyl group has approximate pK values of 9.8–10.4, which are comparable with the pK value of the S0 state of 3-HF (28). The lower affinity binding site of 3-HF, having binding constant of 2.5 × 105 M−1, is situated in the IIA subdomain and is occupied by the normal tautomer. 3-HF molecule bound in this site is located in immediate vicinity of Trp-214, thus serving as a very efficient energy acceptor (energy trap) for the excited tryptophan. Coupling between the Trp-214 → 3-HF normal tautomer intermolecular energy transfer and the intramolecular PT in 3-HF is followed by the PT fluorescence of 3-HF.
An important aspect of the HSA–3-HF interaction derives from the physiological significance of some of flavonols and their analogs. These molecules exhibit not only different excitation modes, but also are known as antioxidants (30) and dietary-derived inhibitors of various types of cancer (31, 32). The flavonols have figured prominently in the new spectroscopic phenomenology, which is the basis of this paper. Intrinsic fluorescence of flavonols provides the unique possibility to monitor their interaction with physiologically important targets in situ. For example, flavonols (3-HF, fisetin, and quercetin) can be used as biologically active fluorescence probes of enzymes involved in carcinogenesis, such as tyrosine kinase (33) and DNA topoisomerase (34).
Acknowledgments
This paper is dedicated to Prof. Michael Kasha, the great scientist and teacher in the field of electronic spectroscopy, on the occasion of his 75th birthday. We are also pleased to acknowledge Prof. Kasha for discussions and suggestions on this manuscript. This research was supported under Contract DE-FG05-87ER60517 between the Office of Health and Environmental Research, U.S. Department of Energy, and Florida State University.
Decomposition of the Absorption Spectra of the HSA–3-HF Complexes into Two Components
The 3-HF absorption of HSA–3-HF complexes represent a sum of absorption bands of the two ionic forms of 3-HF: an anion and a neutral molecule. On the basis of the pH-dependence of the 3-HF absorption (26–28), it is known that the band with λmax = 420 nm corresponds to the anion, while the band with λmax = 340 nm corresponds to the neutral form. The absorption of the neutral form is only weakly dependent on the surrounding medium exhibiting slight spectral shift in different solvents. The absorption spectrum of an ethanol solution of 3-HF, where all the molecules are in the neutral form, was taken for a basis of spectral decomposition of the absorption of HSA–3-HF complexes. Experimentally measured spectra can be represented as:
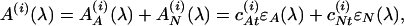 |
4 |
where AA and AN are absorbances of the anion and the neutral form, ɛA and ɛN are corresponding molecular extinction coefficients, and cAt and cNt are total concentrations of each form in the ith solution. The extinction coefficient of the neutral form was taken to be ɛN(λ) = ɛEtOH(λ + Δλ), where ɛEtOH(λ) is the extinction of an ethanol solution and Δλ is a spectral shift to be determined. As follows from Eq. 4:
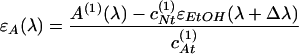 |
5 |
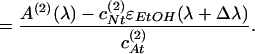 |
A non-linear fitting of parameters cAt, cNt, and Δλ to Eq. 5 was performed using the Marquart–Levenberg algorithm under constraints cAt(i) + cNt(i) = c(i) (i = 1, 2), where c(i) is a total concentration of the probe in the ith solution. This procedure gives the concentrations of each ionic form in each of the solutions as well as the value of the spectral shift Δλ = 2.7 nm. The concentrations of the 3-HF normal tautomer and anion for a solution of 40 μM HSA and 40 μM 3-HF are 16.1 μM and 23.9 μM, respectively, and for a solution of 40 μM HSA and 80 μM 3-HF, they are 41 μM and 39 μM, respectively. Having found the concentrations, we can proceed to determine the binding constants for HSA–3-HF complexes. We will do it using a model taking into account only first two strongest binding sites of HSA and neglecting cooperativity of binding. Within such a model the ratio of concentrations of the two ionic forms bound to protein is:
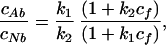 |
6 |
where cf is the concentration of free probe molecules in solution. This ratio varies from k1/k2 for solutions with excess of protein to 1 for solutions with excess of probe. On the other hand, the ratio of concentrations of anion and neutral form bound in the 1:1 protein–probe complex is:
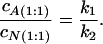 |
These concentrations can be determined as cA(1:1) = cAb − c(1:2) and cN(1:1) = cNb − c(1:2), where c(1:2) is concentration of the 1:2 protein–probe complexes. Assuming noncooperative binding, c(1:2) can be determined as c(1:2) = cNbcAb/cP, where cP is an initial concentration of protein. For large excess of protein, we can neglect both concentration of the 1:2 protein–probe complexes and concentration of free probe and determine the k1/k2 ratio. Absorption measurements on the solutions with large excess of protein over probe (40 μM HSA and 5 μM 3-HF) provide the k1/k2 ratio of 2.8. One can propose that for the HSA–3-HF complexes, cAb = cAt, because there is no free anion in solution at pH = 7.0, while determined concentration of neutral form includes both bound and free molecules: cNt = cNb + cf. We can first determine cNb from the equation:
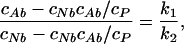 |
and then find cf = cNt − cNb. This procedure gives the cf value of 2.25 μM in the solution of 40 μM HSA and 40 μM 3-HF. Now we can determine the binding constants from Eq. 6, getting values of k1 = 7.2 × 105 M−1 and k2 = 2.5 × 105 M−1.
Determination of the Rate of the Resonant Energy Transfer and the Distance from Trp-214 to 3-HF Anion Bound in the Higher Affinity Binding Site
Within the previously described model, the solution of HSA–3-HF complexes contains four different species relevant for analyzing the fluorescence quenching data. They are:
(i) Free protein with concentration cPf and quantum yield of tryptophan fluorescence:
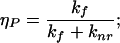 |
(ii) A complex of protein with 3-HF anion bound in the higher affinity binding site with concentration cA(1:1) and quantum yield:
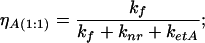 |
(iii) A complex of protein with neutral 3-HF molecule bound in the lower affinity binding site with concentration cN(1:1) and quantum yield:
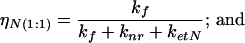 |
(iv) A complex of protein with both ionic forms occupying both binding sites with concentration c(1:2) and quantum yield:
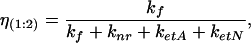 |
where kf is a radiative rate of fluorescence, knr is a total rate of all nonradiation relaxation processes other than the resonance energy transfer, and ketA and ketN are the rates of the resonance energy transfer from Trp-214 to 3-HF anion and neutral molecule, respectively. Because the Trp-214 is one of the hydrophobic residues lining the binding pocket of the lower affinity site, Förster-type energy transfer to a 3-HF molecule located there is very efficient, with ketN ≫ kf and ηN(1:1) ≅ η(1:2) ≅ 0. The concentration of each species can be determined from known concentration of each ionic form bound to protein (see A), assuming noncooperativity of binding as:
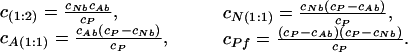 |
Relative fluorescence intensity of a solution of HSA–3-HF complexes in respect to a solution of intact HSA is:
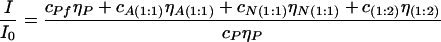 |
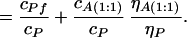 |
7 |
From Eq. 7, we can find ηA(1:1)/ηP and then, using the relation:
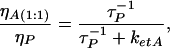 |
where τP is the measured fluorescence lifetime of intact HSA, τP = (kf + knr)−1, we can determine:
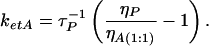 |
In this way we obtain a value of ketA= 1.61 × 108 s−1. According to the Förster theory of resonance energy transfer, this rate is related to intermolecular separation, R, as:
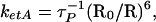 |
where R0 is a Förster radius found from Eq. 3 in Materials and Methods. The value of spectral overlap integral, 1.64 × 10−14 M−1·cm6, was calculated using extinction coefficient of the 3-HF anion, 10,000 M−1·cm−1. The calculated value of Förster radius is 25.4 Å, and estimated distance from Trp-214 to the 3-HF anion situated in the IIIA subdomain is 25.5 Å.
Footnotes
Abbreviations: PT, proton transfer; 3-HF, 3-hydroxyflavone; HSA, human serum albumin.
References
- 1.Marcus R, Sutin N. Biochim Biophys Acta. 1985;811:265–322. [Google Scholar]
- 2.Maroncelli M, MacInnis J, Fleming G R. Science. 1989;243:1674–1681. doi: 10.1126/science.243.4899.1674. [DOI] [PubMed] [Google Scholar]
- 3.Barbara P F, Jarzeba W. Adv Photochem. 1990;15:1–68. [Google Scholar]
- 4.Simon J D. Acc Chem Res. 1988;21:128–134. [Google Scholar]
- 5.Weller A. Z Electrochem. 1956;60:1144–1147. [Google Scholar]
- 6.Kasha M. J Chem Soc Faraday Trans 2. 1986;82:2379–2392. [Google Scholar]
- 7.Saltiel J, D’Agostino J, Megarity E D, Metts L, Neuberger K R, Wrighton M, Zafiriou O. Org Photochem. 1973;3:1–113. [Google Scholar]
- 8.Rothenberger G, Negus D K, Hochstrasser R M. J Chem Phys. 1983;79:5360–5367. [Google Scholar]
- 9.Bañares L, Heikal A A, Zewail A H. J Phys Chem. 1992;96:4127–4130. [Google Scholar]
- 10.Davydov A S. Zh Eksp Teoret Fiz. 1948;18:210–218. [Google Scholar]
- 11.Förster Th. Ann Phys. 1948;2:55–75. [Google Scholar]
- 12.Kasha M, Parthenopoulos D, Dellinger B. Int J Quantum Chem. 1993;45:689–708. [Google Scholar]
- 13.Grabowski Z R, Rotkiewicz K, Siemiarczuk A, Cowley D J, Baumann W. Nouv J Chim. 1979;3:443–454. [Google Scholar]
- 14.Rettig W. Angew Chem Int Ed Engl. 1986;25:971–988. [Google Scholar]
- 15.Heldt J, Gormin D, Kasha M. Chem Phys. 1989;136:321–334. [Google Scholar]
- 16.Chou P-T, Martinez M L, Clements J H. J Phys Chem. 1993;97:2618–2622. [Google Scholar]
- 17.Swinney T C, Kelley D F. J Chem Phys. 1993;99:211–221. [Google Scholar]
- 18.Ormson S M, Brown R G, Vollmer F, Rettig W. J Photochem Photobiol A. 1994;81:65–72. [Google Scholar]
- 19.Sytnik A, Gormin D, Kasha M. Proc Natl Acad Sci USA. 1994;91:11968–11972. doi: 10.1073/pnas.91.25.11968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Förster Th. In: Comparative Effects of Radiation. Burton M, Kirby-Smith J S, Magee J, editors. New York: Wiley; 1960. pp. 300–341. [Google Scholar]
- 21.Kasha M. In: Physical and Chemical Mechanisms in Molecular Radiation Biology. Glass W A, Varma M N, editors. New York: Plenum; 1991. pp. 231–255. [Google Scholar]
- 22.Sengupta P K, Kasha M. Chem Phys Lett. 1979;68:382–385. [Google Scholar]
- 23.Laermer F, Elsaesser T, Kaiser W. Chem Phys Lett. 1988;148:119–124. [Google Scholar]
- 24.Schwartz B J, Peteanu L A, Harris C B. J Phys Chem. 1992;96:3591–3598. [Google Scholar]
- 25.He X M, Carter D C. Nature (London) 1992;358:209–215. doi: 10.1038/358209a0. [DOI] [PubMed] [Google Scholar]
- 26.McMorrow D, Kasha M. J Phys Chem. 1984;88:2235–2243. [Google Scholar]
- 27.Parthenopoulos D A, Kasha M. Chem Phys Lett. 1990;173:303–309. [Google Scholar]
- 28.Wolfbeis O S, Knierzinger A, Schipfer R. J Photochem. 1983;21:67–79. [Google Scholar]
- 29.Eaton D. Pure Appl Chem. 1988;60:1107–1114. [Google Scholar]
- 30.Jovanovic S, Steenken S, Tosic M, Marjanovic B, Simic M G. J Am Chem Soc. 1994;116:4846–4851. [Google Scholar]
- 31.Fotsis T, Pepper M, Adlercreutz H, Fleishmann G, Hase T, Montesano R, Schweigerer L. Proc Natl Acad Sci USA. 1993;90:2690–2694. doi: 10.1073/pnas.90.7.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Messina M J, Persky V, Setchell K D R, Barnes S. Nutr Cancer. 1994;21:113–131. doi: 10.1080/01635589409514310. [DOI] [PubMed] [Google Scholar]
- 33.Van der Geer P, Hunter T, Lindberg R A. Annu Rev Cell Biol. 1994;10:251–337. doi: 10.1146/annurev.cb.10.110194.001343. [DOI] [PubMed] [Google Scholar]
- 34.Wigley D B. Annu Rev Biophys Biomol Struct. 1995;24:185–208. doi: 10.1146/annurev.bb.24.060195.001153. [DOI] [PubMed] [Google Scholar]