

Abstract
The β-adrenergic receptor kinase 1 (βARK1) is a member of the G protein-coupled receptor kinase (GRK) family that mediates the agonist-dependent phosphorylation and desensitization of G protein-coupled receptors. We have cloned and disrupted the βARK1 gene in mice by homologous recombination. No homozygote βARK1−/− embryos survive beyond gestational day 15.5. Prior to gestational day 15.5, βARK1−/− embryos display pronounced hypoplasia of the ventricular myocardium essentially identical to the “thin myocardium syndrome” observed upon gene inactivation of several transcription factors (RXRα, N-myc, TEF-1, WT-1). Lethality in βARK1−/− embryos is likely due to heart failure as they exhibit a >70% decrease in cardiac ejection fraction determined by direct in utero intravital microscopy. These results along with the virtual absence of endogenous GRK activity in βARK1−/− embryos demonstrate that βARK1 appears to be the predominant GRK in early embryogenesis and that it plays a fundamental role in cardiac development.
Keywords: gene cloning, knock out, phosphorylation, thin myocardium syndrome
G protein-coupled receptors are regulated by multiple mechanisms that serve to adapt their signaling ability to a constantly changing environment. One of the most rapid mechanisms modulating agonist-activated receptors is phosphorylation by G protein-coupled receptor kinases (GRKs) resulting in the uncoupling of the G protein-mediated transduction process (1). This phenomenon, termed desensitization, affects both sensory and hormonally responsive systems. Six distinct GRKs (GRK1-6) have been identified to date. GRK1 or rhodopsin kinase, and GRK2 or β-adrenergic receptor kinase 1 (βARK1), are the best studied members of the family (1, 2). Phosphorylation of G protein-coupled receptors by GRKs increases the receptor affinity for cytosolic proteins named arrestins, which mediate uncoupling of the receptor from G proteins, leading to transient desensitization (3) followed in time by recovery of responsiveness (4).
Because of the lack of GRK selective drugs, the physiological function of βARK1 is still largely unknown. With the exception of the visual system in rodents and fly, where rhodopsin kinase and arrestins have been implicated in the quenching of the light reaction (5, 6), relatively little is known about the role and function of GRKs in vivo. Initial insights into the potential in vivo function of mammalian βARK1 have recently come from investigating the potential role of βARK1 activity in the heart where β-adrenergic receptors regulate cardiac chronotropy and inotropy (7). Transgenic mice over-expressing either βARK1 or a peptide inhibitor of βARK1 in the heart demonstrated that the activity of this kinase significantly contributes to the regulation of myocardial contractility (8).
To gain further insights into the role of βARK1 in vivo, and to determine its physiological involvement in the regulation of G protein-coupled receptor function, we have disrupted the βARK1 gene in mice. Our results reveal the unexpected finding that βARK1 is required for embryonic cardiac development and function and suggest a possible convergence between G protein-coupled receptor signal transduction and transcription factor pathways with respect to cardiac development.
MATERIALS AND METHODS
βARK1 Gene Inactivation Procedure.
For construction of the targeting vector and production of chimeric mice, a 5.4-kb HindIII–EcoRI fragment comprising exons 9–21 of the βARK1 gene was subcloned in HindIII–EcoRI of pGEM4Z (Promega) to generate plasmid pBK2-8HE. This plasmid was linearized with HindIII, and an adapter (NotI, XbaI, EcoRI, SalI, HindIII) was inserted to produce pBK2-8AHE. The neomycin resistance cassette was excised by XhoI and EcoRI from the plasmid pD383 (gift from R. Hen, Columbia University, New York), where this gene is under the control of the human phosphoglycerate kinase promoter, followed by the phosphoglycerate kinase poly(A+) signal. An 1822-bp fragment was amplified by PCR between a primer 3′ of exon 4 (CACATGAATTCACACCCCACAGGACTCTTAC, EcoRI) and a primer 3′ of exon 2 (GCTCAGTCTAGAGGGAAAACTTGGGGTG, XbaI). This XbaI–EcoRI fragment was directionally subcloned, with the Neo cassette, in pBK2-8AHE digested by XbaI and SalI, to generate the targeting plasmid pBK-EHA14/Neo. Before electroporation of embryonic stem cells, pBK-EHA14/Neo was linearized by NotI. Electroporation of embryonic stem cells, cell culture conditions, G418 selection, DNA preparation, and generation of chimeric mice were as described (9). Following transfection of the targeting construct in embryonic stem cells, 108 clones were tested by Southern blotting after digestion with either EcoRI or XbaI. Five clones were found positive for one event of homologous recombination. Two clones were further karyotyped and used for blastocyst injections. Numerous chimeric males derived from EP-127.17 and EP-127.23 tested positive for germ-line transmission. The results presented here correspond to the germ-line EP-127.23.
Southern Blot Procedure.
Southern blots were performed as described (9). Probe A was a 1.1-kb cDNA obtained by PCR amplification in the intron located 5′ of exon 2 using the primer pair 5′-CAGGTTGGCAGGTCTAGGG and 5′-TCTACAGAGGGAGTTCCAGG. DNA was isolated from the tail of adults and from tail and limbs of embryos following an overnight digestion with proteinase K. Samples of DNA were digested with excess of EcoRI, electrophoresed through 0.8% agarose gels, and transferred to nitrocellulose membranes prior to hybridization. Filters were washed at decreasing concentrations of SSC with the final wash being in 0.1× SSC/0.1% SDS at 65°C and exposed to X-R Kodak film for 48 hr.
Analysis of Embryos.
For developmental studies βARK1+/− females were put in the presence of βARK1+/− males for 3 hr and were checked for plugs. Positively plugged females were considered at that point at gestational day 0 (E0). Embryos were dissected at various time points (E7.5 on) and genotyped as above. For Northern blot studies, the rest of the embryo was frozen in dry ice and kept at −80°C until further use. For histological procedures, embryos were put in formalin for at least 24 hr, then embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin following standard procedures.
Gene Expression Studies.
RNA from E13 embryos (obtained from βARK1 heterozygous crossings) was extracted and poly(A)+ RNA was isolated using oligo-d(T) (Amersham). Poly(A)+ RNA (20 μg), originating from embryos of each genotype, was run on a 1.2% agarose gel and transferred to a nylon membrane as described (10). For protein immunoblotting, embryonic or myocardial extracts were prepared as described (8). Equal amounts of protein were then electrophoresed on SDS/12% polyacrylamide gels and transferred to nitrocellulose. βARK1 was then identified by standard Western blotting techniques using polyclonal antisera raised to the carboxyl terminus of βARK1 and chemiluminescent detection as described (8).
Assessment of GRK Activity.
Concentrated supernatants from adult heart and whole embryo extracts were used to phosphorylate rhodopsin-enriched bovine rod outer segment membranes as described (8). Extracts (50 μg protein) were incubated in 75 μl with rod outer segment membranes in lysis buffer with 10 mM MgCl2 and 0.1 mM ATP (containing [γ-32P]ATP). Purified Gβγ was added to some samples for maximal activity of βARK1. After incubation in white light for 15 min, the reactions were quenched with 300 μl of cold lysis buffer and centrifuged for 15 min at 13,000 × g. The sedimented protein was then resuspended in 20 μl of lysis buffer/protein dye and electrophoresed on SDS/12% polyacrylamide gels. Phosphorylated rhodopsin was quantified on a Molecular Dynamics PhosphorImager.
Determination of Cardiac Ejection Fraction in Embryos.
At E12.5–13.5 pregnant mice were anesthetized with ketamine (100 mg/kg i.p.) and xylazine (2.5 mg/kg i.p.) and were intubated and ventilated as described (11, 12). Embryos were visualized by intravital microscopy as described (13) with the exception that real images obtained with a color CCD camera were analyzed instead of fluorescent images. End-diastolic and end-systolic frames defined as the largest and the smallest images during the cardiac cycle were digitized and analyzed. The area and the long axis of each left ventricle (LV) image were measured with National Institutes of Health image software. The border of each blood pool image was traced three times and averaged using the single-plane area-length method (14). End-diastolic and end-systolic LV volumes were calculated using the following equation V = (8/3)(1/π)(1/L)(A)2, where V is LV volume, A is the traced LV area, and L is the long axis measured directly from the digitized video image. The LV ejection fraction was calculated from the left ventricular end-diastolic (LVEDV) and end-systolic volume (LVESV) as (LVEDV − LVESV)/LVEDV.
RESULTS
βARK1 Gene Disruption Is Lethal at Early Gestational Age.
The catalytic subdomain I, which forms part of the ATP binding site, is encoded by exon 8. This exon was removed, together with exons 5, 6, and 7, in the targeting vector for the gene inactivation procedure to ensure that the resulting construct did not confer any enzymatic activity (Fig. 1). Furthermore, the deletion altered the remaining reading frame. A Southern blot of EcoRI-digested genomic DNA extracted from tails of 3-week old litters, which were obtained by crossing of βARK1 heterozygotes, is shown in Fig. 2 Upper. Wild-type DNA gives a band at 25 kb, whereas the mutant genotype gives a band at 17.5 kb as schematically represented in Fig. 1. None of the 623 offspring examined from these crossings resulted in βARK1−/− genotype indicating that the homozygotes die during gestation (Table 1). The ratio of adult heterozygotes to wild type is 2:1 and is consistent with Mendelian segregation when homozygotes are lethal and heterozygotes are viable (Table 1). βARK1+/− mice appear normal with no difference in body weight or size at all ages examined (from birth until 9 weeks of age) (data not shown). Embryos from E7.5 to birth were genotyped (Fig. 2 Lower, Table 1). At E7.5 very little resorption is observed. Necrotic βARK1−/− embryos are found from E9 on, suggesting that the lethality of βARK1−/− embryos begins at this time. Of the viable embryos, the proportion of βARK1−/− embryos is roughly 25% at mid-gestation days and decreases progressively up to E15.5 (Table 1). Following that gestational time, no viable βARK1−/− embryos are ever observed.
Figure 1.
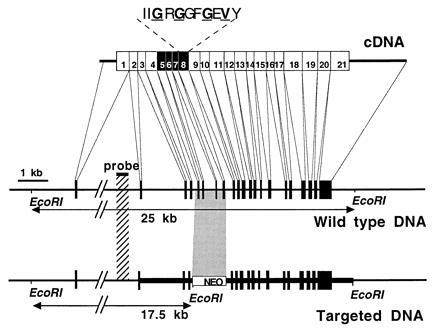
Targeted disruption of the βARK1 gene. (Top) βARK1 cDNA and location of the 21 exons. Screening of a mouse sv129 genomic library (gift from H. S. Kim, University of North Carolina, Chapel Hill) with two probes obtained from either rat βARK1 or βARK2 cDNAs identified six clones of 12–15 kb. These clones were overlapping as evidenced by restriction analysis and Southern blotting (data not shown) and reconstituted a 23-kb DNA fragment. Exons 5–8 (solid boxes) were removed in the final construct for the gene inactivation. Exon 8 contains the consensus catalytic subdomain I of the protein kinases (15). The Gly and Val residues, in boldface type and underlined, are known to bind ATP. (Middle) Physical map of the βARK1 gene in the wild-type mouse. The thin line represents the introns in the genomic DNA, and solid boxes represent the exons. Intron 1 (>9 kb) is interrupted, as its actual size was not determined. The EcoRI restriction fragment length is determined on the basis of agarose gel electrophoresis and Southern blotting using the denoted probe. (Bottom) Physical map of the βARK1 gene in mouse disrupted by homologous recombination. The thick line represents the extent of the targeting construct. The counterpart of the inserted neo resistance DNA cassette is indicated by a shaded box. Solid boxes denote the exons, and an additional EcoRI site generating a 17.5-kb restriction fragment is represented.
Figure 2.
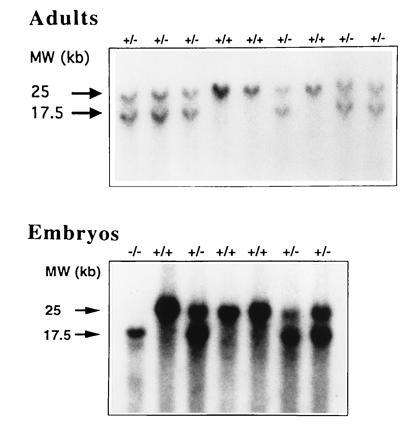
Southern blot analysis of EcoRI-digested genomic DNA following the crossing of male and female heterozygote mice. DNA was extracted from (Upper) tails of 3-week-old littermates or (Lower) tails and limbs of littermate embryos at E13. Blots were hybridized with a 5′ flanking probe. Wild-type and mutant allele correspond to 25 and 17.5 kb fragments, respectively.
Table 1.
Genotypic characterization of offspring from the βARK1 gene disruption
| Offspring | Measurement | Homozygous | Heterozygous | Wild type | Total |
|---|---|---|---|---|---|
| E9–E15.5 | No. of offspring | 39 | 85 | 48 | 172 |
| % actual | 23 | 49 | 25 | 100 | |
| (% theoretical) | (25) | (50) | (25) | (100) | |
| E16–E20 | No. of offspring | 0 | 39 | 18 | 57 |
| % actual | 0 | 70 | 30 | 100 | |
| (% theoretical) | (0) | (67) | (33) | (100) | |
| Postnatal | No. of offspring | 0 | 402 | 221 | 623 |
| % actual | 0 | 64 | 36 | 100 | |
| (% theoretical) | (0) | (67) | (33) | (100) |
Heterozygote mice were crossed and the offspring’s genotype was analyzed during ontogeny and after birth as described in Materials and Methods. E, gestational day.
βARK1 Is the Major Kinase During Development.
To demonstrate that the embryonic lethality is associated with a lack of functional βARK1 and that the disruption of the βARK1 gene does indeed produce the expected functional ablation of the enzyme, biochemical characterization including Northern and Western blots, as well as GRK activity measurements were performed on E11-E13 embryos (Fig. 3). Northern blot analysis shows that βARK1 mRNA is expressed very early during development and that this expression is very low since analysis had to be performed on 20 μg of poly(A)+ RNA with prolonged exposure of the blots. Two bands are obtained for the βARK1 transcripts from both wild-type adult and embryo specimens as previously described for extracts of adult bovine (15) or human tissues (16) (Fig. 3a). βARK1+/− embryos show a hybridization signal of markedly reduced intensity (62% decrease). No signal is obtained in βARK1−/− embryos (Fig. 3a). The pattern of protein expression for βARK1 correlates well with mRNA levels in each genotype, i.e., early embryonic expression of a protein of the expected 80 kDa size, reduced expression in the βARK1+/− embryos, and no signal in the βARK1−/− littermates (Fig. 3b).
Figure 3.
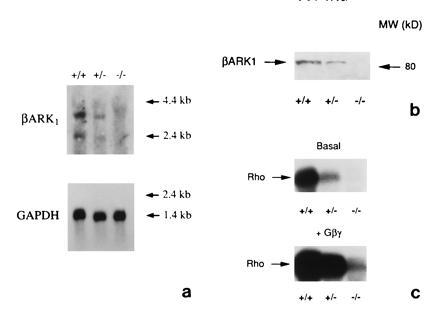
Biochemical characterization of the effects of βARK1 gene disruption on extracts from E13 embryos obtained after crossing of heterozygote mice. (a) Northern blot studies on 20 μg of poly(A)+ RNA extracted from whole E13 embryos hybridized with a cDNA probe for βARK1 (Upper) or glyceraldehyde-3-phosphate dehydrogenase (Lower) used as control. (b) Western blot analysis using a rabbit polyclonal antiserum to the COOH terminus of βARK1 (8). Chemiluminescent detection of alkaline phosphatase-conjugated secondary antibody was used. (c) Assessment of GRK activity against rhodopsin (see Materials and Methods).
To determine the GRK activity in relation to the embryo’s genotype, we assessed whole embryo extracts for the ability to phosphorylate light-activated rhodopsin, a prototypic G protein-coupled receptor and an in vitro substrate for βARK1 (8). G protein βγ subunits were also added to the reaction to achieve maximum levels of rhodopsin phosphorylation. βγ subunits of heterotrimeric G proteins target βARK1 to the membrane, which presumably facilitates the precise orchestration of βARK1 phosphorylation of only activated receptors. Under the conditions of our experiments, most GRKs can phosphorylate rhodopsin. Extracts from wild-type embryos show a significant level of rhodopsin phosphorylation, which is increased upon addition of the βγ subunits. Extracts from heterozygote embryos show lower levels of activity, which correlate with the decrease in βARK1 mRNA and protein. In contrast to wild-type animals, extracts from E13 homozygote embryos are virtually devoid of activity (Fig. 3c). Even in the presence of βγ subunits, only a faint rhodopsin phosphorylation signal (<1%) is observed. Although we cannot exclude the possibility that other GRKs are expressed at very low levels, Western blots of early embryonic extracts reveal no detectable protein expression of other prominent members of the GRK family such as GRK3 (βARK2) and GRK5 (data not shown). This is consistent with the idea that no other GRK is expressed at levels high enough to produce either significant rhodopsin phosphorylation or compensate for βARK1 gene disruption in early development.
βARK1−/− Embryos Show Myocardial Hypoplasia.
Anatomical examination of βARK1−/− embryos reveals that they are significantly smaller in size, with an overall development retarded by approximately 24–36 hr at E15.5 in comparison to wild-type littermates. They have a generally paler appearance than their littermate controls and heterozygotes. This is suggestive of insufficient vascular irrigation and not of a hematopoietic deficiency since the liver appears normal. Obvious cardiac abnormalities in βARK1−/− embryos are found from E11 on. Examination of dissected hearts at E13 reveals that the right atrium of βARK1−/− embryos is significantly enlarged as compared with littermate controls (data not shown). Histological sections of stained paraffin-embedded embryos reveal striking abnormalities in the myocardium of the βARK1−/− embryos, which are reminiscent of the “thin myocardium syndrome” observed with the targeted inactivation of several transcription factors in the mouse (18) (Fig. 4). Hypoplasia of both the right and left ventricular myocardium is apparent, with notable hypoplasia in the right and left atria, and remarkable hypoplasia and dysplasia of the interventricular septum. In some of the embryos, the nontrabeculated ventricular myocardium consists of only a single cell layer as shown in Fig. 4. The ventricular and atrial cavities (lumina) appear unusually large as a consequence of the hypoplasia. There is persistence of a wide interventricular foramen in the βARK1−/− embryos. The interventricular foramen normally persists in the mouse embryo until E14–E15 when the interventricular septum fuses with the conotruncal ridges, dividing the right and left ventricles (19). The persistence of an unusually wide interventricular canal in the βARK1−/− embryos might be attributable to its retarded growth. However, the dysplasia and hypoplasia of the myocardium do not reflect the condition of a younger heart, but represent a true and profound dysmorphogenesis. This is evidenced by the abnormally thin compact myocardial layer, the numerous and thinned trabeculae, and the lack of cellular density where the muscular interventricular septum should be forming. The endocardial and epicardial layers appear normal. Slight variation in the extent or severity of the defect are observed among embryos at different gestational stages. This observation may be the consequence of the lack of animals of pure genetic background and account for the variation in the time of death of the embryos (E9.5–E15.5) (20).
Figure 4.
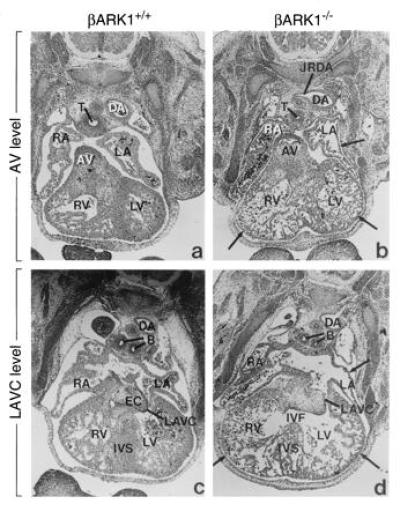
Cardiac abnormalities in βARK1 homozygote embryos visualized with hematoxylin- and eosin-stained sections of whole embryos. Two sections each are shown at the aortic valve and left atrioventricular canal level. Structures are labeled as follows: B, bronchi; DA, descending aorta; EC, endocardial cushion; IVF, interventricular foramen; IVS, interventricular septum; JRDA, junction of the right dorsal aorta; LA, left atrium; LAVC, left atrioventricular canal; LV, left ventricle; RA, right atrium; RV, right ventricle; T, trachea.
The reduced thickness of the compact layer of the myocardium and the unusually thinned trabeculae in the ventricles and atria of βARK1−/− embryos suggest a role for βARK1 in the normal migration, differentiation, or proliferation of myocardial cells from splanchnic mesoderm (originating in the precardiac mesoderm). However, the normal morphology of the endocardial cushions and epicardium (also derived from splanchnic mesoderm of the precardiac region) suggests that it is only the sub-population of the precardiac mesoderm giving rise to the compact myocardium and trabeculated myocardium that are, directly or indirectly, dependent upon βARK1 gene expression. Moreover, the normal appearance of cardiac structures containing neural crest cells (conotruncus, aortic sac, and great vessels of the heart) in the βARK1−/− embryos suggests that the βARK1 gene is not essential for normal migration of the neural crest into the walls of the aortico-pulmonary outflow tract (18).
βARK1−/− Embryos Die from Heart Failure.
Analysis on E12.5–E13.5 embryos demonstrates significant impairment in heart function of homozygote embryos in comparison to littermate wild-type embryos, which correlates closely with the histological findings. Intravital microscopy in utero with direct visualization of intracardiac blood pools reveals a marked decrease in the cardiac ejection fraction of homozygote embryos, suggesting heart failure as a probable cause of lethality. Indeed, whereas the calculated LV ejection fraction in wild-type hearts (n = 6) averages 56%, that of βARK1−/− embryos (n = 5) is 16%, with some of them having an ejection fraction as low as 9% (Fig. 5). These results provide direct evidence that the thin myocardium syndrome observed in these animals results in ventricular chamber dysfunction of a magnitude sufficient to cause embryonic heart failure. Interestingly, a similar impairment in cardiac function has recently been shown in mice carrying a similar morphological syndrome due to inactivation of the RXRα gene for retinoic acid (21).
Figure 5.
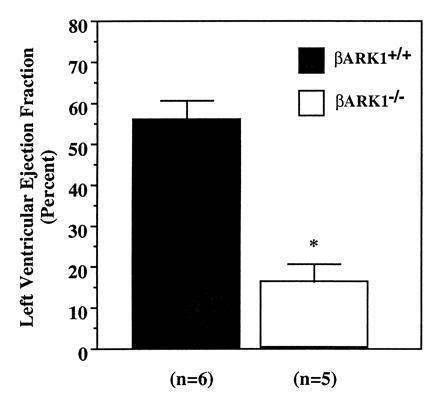
In vivo assessment of cardiac ejection fraction in E12.5–E13.5 embryos. Images were obtained on embryos with an intravital microscope and recorded at 30 frames/sec. n = the number of embryos from three different litters.
DISCUSSION
In this work, we describe the characteristics of the genetic disruption of the mouse βARK1 (GRK2) gene, a member of the GRK family. Our data strongly suggest that (i) βARK1 appears to be the major contributor to the GRK activity present at this stage of development, (ii) that it is crucial during embryogenesis, and (iii) that it plays a key role in cardiac development and function as evidenced by the appearance of a thin myocardium syndrome, cardiac pump failure, and fetal death of βARK1−/− embryos. Thus, our results point to βARK1 as an essential protein during development such that no other GRK can compensate for its loss.
The finding that βARK1 disruption dramatically impairs cardiac development and function during early embryogenesis despite low levels of expression of the corresponding mRNA in normal adult heart (data not shown) and whole embryos (Fig. 3a) is unexpected. βARK1−/− embryos exhibit a pronounced hypoplasia of the ventricular myocardium and an obvious lack of organization or differentiation of the ventricular myocardium. This phenotype is observed between E12.5–E15.5 when one major cause of embryonic lethality can be inefficient pumping action of the heart leading to circulatory failure. Indeed, the LV ejection fraction of homozygote embryos measured in utero is 29% that of wild-type littermates. This strongly suggests that homozygote embryos are dying from heart failure as a consequence of an abnormally thin compact layer and disorganized trabeculae in the ventricular wall.
The proper functioning of the cardiovascular system is a prerequisite for embryonic survival. A large portion of preterm abortions can presumably be accounted for by congenital circulatory system abnormalities (18). Consequently, understanding the mechanisms and factors involved in cardiogenesis is of critical importance. Cardiac specific expression of genes occurs between E8 and E16 (22). This correlates with the differentiation, regionalization, septation, and growth of the developing heart. Cardiac cell proliferation and differentiation is also controlled in part by paracrine and autocrine mechanisms including hormones and growth factors such as norepinephrine, endothelin-1, and angiotensin, which activate G protein-coupled receptors (23, 24). Through G protein-coupled receptor dependent mechanisms catecholamines control chronotropy and inotropy in the normally functioning heart. Evidence suggests that the regulation of these signaling events through GRKs plays a critical role in the response to adrenergic agents and may be modified in heart failure (25). Thus, the essential role of βARK1 in cardiogenesis suggests that it may represent a candidate gene susceptible to congenital mutations, which lead to fetal death.
Interestingly, inactivation of the βARK1 gene leads to precisely the same cardiac developmental defects that are seen with inactivation of several transcription factor genes such as RXRα (26, 27), N-myc (28), TEF-1 (29), and WT-1 (30). This suggests that, during development, these transcription factors or processes under their control may be regulated by as yet unidentified G protein-coupled receptor signaling pathways, the function of which is distorted by the absence of the βARK1 regulatory system. It is known, for example, that G protein-coupled receptors modulate the activity of several transcription factors via p21ras-dependent and -independent signaling and MAP kinase cascades (31). Second messengers such as cAMP, the levels of which are regulated by G protein-coupled receptors, are also known to regulate several transcription factor pathways (31). Moreover, it has been shown in neonatal cardiomyocytes that retinoic acid inhibits G protein-coupled receptor-mediated endothelin and α-adrenergic signals leading to cardiac hypertrophy (32). Therefore, it is conceivable that during embryonic development the same or different signals mediated through G protein-coupled receptors and affected by retinoic acid might serve to program maturation of ventricular myocytes. Alternatively, the effects of βARK1 gene disruption may result from the interaction of GRKs with components of other cellular processes. The identification of a potential convergence between G protein-coupled receptor signaling and transcription factors in cardiac development may provide an important framework for the characterization of the signals and determinants of cardiogenesis. In summary, the results presented here suggest that βARK1 (GRK2) plays a fundamental role during development and thus participates in intracellular signal transduction mechanisms, which regulate cardiogenesis.
Acknowledgments
We would like to thank Dr. B. H. Koller for the efficient production of embryonic stem cells and chimeric mice, Dr. D. Severynse for expert advice in manipulating transgenic animals, and Drs. L. Barak, A. Macrae, S. Ferguson, M. Kirby, and R. Premont for helpful discussion. We also thank N. Bhargava for his help in analysis of embryonic function and S. Suter for her expert technical help. Special thanks go to Dr. H. M. Sucov for his critical review of the manuscript. M.J. was a recipient of a long-term European Molecular Biology Organization fellowship. B.G. was a recipient of a National Alliance for Research on Schizophrenia and Depression young researcher fellowship. M.G.C. is the recipient of a Bristol–Myers Squibb Unrestricted Neuroscience Award. This work was supported in part by National Institutes of Health Grants NS 19576 (M.G.C.) and HL 16037 (R.J.L.), and American Heart Asociation Texas affiliate Grant 96G-458 (R.A.B.).
Footnotes
Abbreviations: βARK, β-adrenergic receptor kinase; GRK, G protein-coupled receptor kinase; E, gestational day; LV, left ventricle.
References
- 1.Lefkowitz R J, Caron M G. Handb Exp Pharmacol. 1993;46:33–42. [Google Scholar]
- 2.Inglese J, Freedman N, Koch W, Lefkowitz R J. J Biol Chem. 1993;268:23735–23738. [PubMed] [Google Scholar]
- 3.Lohse M J, Andexiner S, Pitcher J, Trukawinski S, Codina J, Faure J P, Caron M G, Lefkowitz R J. J Biol Chem. 1992;267:8558–8564. [PubMed] [Google Scholar]
- 4.Ferguson S S G, Downey W E, III, Colapietro A M, Barak L, Menard L, Caron M G. Science. 1996;271:363–366. doi: 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Makino C L, Peachey N S, Baylor D A, Simon M I. Science. 1995;267:374–377. doi: 10.1126/science.7824934. [DOI] [PubMed] [Google Scholar]
- 6.Dolph P J, Ranagamathan R, Colley N J, Hardy R W, Socolich M, Zuker C S. Science. 1993;260:1910–1916. doi: 10.1126/science.8316831. [DOI] [PubMed] [Google Scholar]
- 7.Lohse M J. Trends Cardiovasc Med. 1995;5:63–68. doi: 10.1016/1050-1738(94)00034-4. [DOI] [PubMed] [Google Scholar]
- 8.Koch W J, Rockman H A, Samama P, Hamilton R, Bond R, Milano C A, Lefkowitz R. Science. 1995;268:1350–1353. doi: 10.1126/science.7761854. [DOI] [PubMed] [Google Scholar]
- 9.Koller B H, Smithies O. Proc Natl Acad Sci USA. 1989;86:8932–8935. doi: 10.1073/pnas.86.22.8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaber M, Fournier M C, Bloch B. Mol Brain Res. 1992;15:189–194. doi: 10.1016/0169-328x(92)90108-n. [DOI] [PubMed] [Google Scholar]
- 11.Rockman H A, Ross R S, Harris A N, Knowlton K U, Steinhelper M E, Field L J, Ross J, Jr, Chien K R. Proc Natl Acad Sci USA. 1991;88:8277–8281. doi: 10.1073/pnas.88.18.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rockman H A, Ono S, Ross R S, Jones L R, Karimi M, Bhargava V, Ross J, Jr, Chien K R. Proc Natl Acad Sci USA. 1994;91:2694–2698. doi: 10.1073/pnas.91.7.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dyson E, Sucov H M, Kubalak S W, Schmid-Schönbein G W, DeLano F A, Evans R M, Ross J, Jr, Chien K, R. Proc Natl Acad Sci USA. 1995;92:7386–7390. doi: 10.1073/pnas.92.16.7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandler H, Dodge H T. Am Heart J. 1968;75:325–334. doi: 10.1016/0002-8703(68)90089-6. [DOI] [PubMed] [Google Scholar]
- 15.Hanks S K, Quinn A M, Hunter T. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 16.Benovic J L, DeBlasi A, Stone C W, Caron M G, Lefkowitz R J. Science. 1989;246:235–240. doi: 10.1126/science.2552582. [DOI] [PubMed] [Google Scholar]
- 17.Chuang T T, Sallese M, Ambrosini G, Parruti G, De Blasi A. J Biol Chem. 1992;267:6886–6892. [PubMed] [Google Scholar]
- 18.Rossant J. Circ Res. 1996;78:349–353. doi: 10.1161/01.res.78.3.349. [DOI] [PubMed] [Google Scholar]
- 19.Vuillemin M, Pexieder T. Am J Anat. 1989;184:114–128. doi: 10.1002/aja.1001840203. [DOI] [PubMed] [Google Scholar]
- 20.Threadgill D W, Dlugosz A A, Hansen L A, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris R C, Barnard J A, Yuspa S H, Coffey R J, Magnuson T. Science. 1996;269:230–234. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- 21.Dyson E, Sucov H M, Kubalak S W, Schmid-Schonbein G N, DeLano F A, Evans R M, Ross J, Jr, Chien K R. Proc Natl Acad Sci USA. 1995;92:7386–7390. doi: 10.1073/pnas.92.16.7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kubalak S W, Miller-Hance W C, O’Brien T X, Dyson E, Chien K R. J Biol Chem. 1994;269:16961–16970. [PubMed] [Google Scholar]
- 23.Chien K R, Knowlton K U, Zhu H, Chien S. FASEB J. 1991;5:3037–3046. doi: 10.1096/fasebj.5.15.1835945. [DOI] [PubMed] [Google Scholar]
- 24.Chien K R, Zhu H, Knowlton K U, Miller-Hance W, van Bilson M, O’Brien T X, Evans S M. Annu Rev Physiol. 1993;55:77–97. doi: 10.1146/annurev.ph.55.030193.000453. [DOI] [PubMed] [Google Scholar]
- 25.Ungerer M, Parruti G, Böhm M, Puzicha M, DeBlasi A, Erdmann E, Lohse M J. Circ Res. 1994;74:206–213. doi: 10.1161/01.res.74.2.206. [DOI] [PubMed] [Google Scholar]
- 26.Sucov H M, Dyson E, Gumeringer C I, Price J, Chien K R. Genes Dev. 1994;8:1007–1018. doi: 10.1101/gad.8.9.1007. [DOI] [PubMed] [Google Scholar]
- 27.Kastner P, Grondona J M, Mark M, Gansmuller A, LeMeur M, Decimo D, Vonesch J-L, Dolle P, Chambon P. Cell. 1994;78:987–1003. doi: 10.1016/0092-8674(94)90274-7. [DOI] [PubMed] [Google Scholar]
- 28.Charron J, Malynn B A, Fisher P, Stewart V, Jeanotte L, Goff S P, Robertson E J, Alt F W. Genes Dev. 1992;6:2248–2257. doi: 10.1101/gad.6.12a.2248. [DOI] [PubMed] [Google Scholar]
- 29.Moens C B, Stanton B R, Parada L F, Rossant J. Development (Cambridge, UK) 1993;119:485–499. doi: 10.1242/dev.119.2.485. [DOI] [PubMed] [Google Scholar]
- 30.Kreidberg J A, Sariola H, Loring J M, Maeda M, Pelletier J, Housman D, Jaenisch R. Cell. 1993;74:679–691. doi: 10.1016/0092-8674(93)90515-r. [DOI] [PubMed] [Google Scholar]
- 31.Dhanasekaran N, Heasly L E, Johnson G L. Endocr Rev. 1995;16:259–270. doi: 10.1210/edrv-16-3-259. [DOI] [PubMed] [Google Scholar]
- 32.Zhou M D, Sucov H M, Evans R M, Chien K R. Proc Natl Acad Sci USA. 1995;92:7391–7395. doi: 10.1073/pnas.92.16.7391. [DOI] [PMC free article] [PubMed] [Google Scholar]