

Abstract
In the current model for bacterial cell division, FtsZ protein forms a ring that marks the division plane, creating a cytoskeletal framework for the subsequent action of other proteins such as FtsA. This putative protein complex ultimately generates the division septum. Herein we report that FtsZ and FtsA proteins tagged with green fluorescent protein (GFP) colocalize to division-site ring-like structures in living bacterial cells in a visible space between the segregated nucleoids. Cells with higher levels of FtsZ–GFP or with FtsA–GFP plus excess wild-type FtsZ were inhibited for cell division and often exhibited bright fluorescent spiral tubules that spanned the length of the filamentous cells. This suggests that FtsZ may switch from a septation-competent localized ring to an unlocalized spiral under some conditions and that FtsA can bind to FtsZ in both conformations. FtsZ–GFP also formed nonproductive but localized aggregates at a higher concentration that could represent FtsZ nucleation sites. The general domain structure of FtsZ–GFP resembles that of tubulin, since the C terminus of FtsZ is not required for polymerization but may regulate polymerization state. The N-terminal portion of Rhizobium FtsZ polymerized in Escherichia coli and appeared to copolymerize with E. coli FtsZ, suggesting a degree of interspecies functional conservation. Analysis of several deletions of FtsA–GFP suggests that multiple segments of FtsA are important for its localization to the FtsZ ring.
One of the major puzzles in cell biology is how proteins involved in cytokinesis are targeted to a the point in a cell, usually the middle, between the daughter genomes (1, 2). In prokaryotes, the essential cell division protein FtsZ localizes to the cell midpoint very early in cytokinesis (3, 4). The mechanism of timing and localization is unknown. FtsZ is probably present in all eubacteria, archaea, and chloroplasts (5–7). FtsZ is highly abundant, with about 5000–20,000 monomers per Escherichia coli cell during exponential growth (8). Immunoelectron microscopy of thin sections of bacterial cells demonstrated that FtsZ polymerizes to form a circumferential ring at the mid-cell division site, constricting at the leading edge of the invaginating septum that eventually separates the two daughter cells (9, 10). The FtsZ ring is thus reminiscent of both the septin and actin rings formed at the division site of animal and yeast cells (2, 11) and the microtubule-based preprophase band of plant cells (12).
Consistent with its cytoskeletal role, FtsZ has certain properties in common with the eukaryotic cytoskeletal protein tubulin. Like tubulin, purified FtsZ binds and hydrolyzes GTP and polymerizes to form long tubules in a GTP-dependent manner (13–17). Recently, it has been shown that purified FtsZ polymerizes into structures that closely resemble those formed by purified tubulin (18). Also, molecular phylogenetic data indicates that an archaeal FtsZ is the most tubulin-like of all prokaryotic FtsZ proteins found so far, suggesting that tubulin may have evolved from an ancestral FtsZ (6).
In addition to FtsZ, there is a growing list of other proteins that are essential for cell division in E. coli. Genetic data and recent biochemical data suggest that these proteins may form a membrane-associated complex with the FtsZ ring (19). One protein, FtsA, is a member of a large family of ATPases that includes actin and Hsp70 (20, 21). It is often localized to the cytoplasmic membrane (22) and acts at a later stage of septation than FtsZ (23, 24), but its role in cell division is unknown. The amount of FtsA is much lower than that of FtsZ, 50–200 molecules per cell (25). When FtsZ is overproduced at least 10-fold above wild-type levels, cell division is inhibited as effectively as when FtsZ is inactivated (26). However, this inhibition can be suppressed by simultaneous overproduction of FtsA, suggesting that FtsZ–FtsA stoichiometric balance is crucial and that both proteins interact (8, 27). Similarly, overproduction of FtsA inhibits cell division and induces large bulges at septation sites, perhaps due to disruption of the septal peptidoglycan breakage and synthesis machinery (28, 29).
In this paper we use green fluorescent protein (GFP) from Aequorea victoria (30, 31) to tag FtsZ and FtsA so that their structure and location can be visualized in living E. coli cells. GFP is a highly fluorescent protein with a self-contained fluorophore and no known external substrate requirements other than molecular oxygen (32). It has become a valuable tool in probing localization of proteins within living cells of many species (33) including bacteria (34). Our results suggest that GFP will be a useful reagent for understanding the dynamics of bacterial cell division.
MATERIALS AND METHODS
Bacterial Strains and Media.
Strains JM109 (recA) or JM105 (Rec+) that contain lacIq on an F′ and MC1061 were used as wild-type E. coli host strains (35). Temperature-sensitive mutant strains include JFL101 (obtained from J. Lutkenhaus, University of Kansas Medical Center), which contains the ftsZ84 allele (36), and strain AX621 containing ftsA1882 (37) (obtained from the E. coli Genetic Stock Center). The standard growth medium was Luria–Bertani (LB) medium or agar (35) containing 0.5% NaCl instead of 1% NaCl; it was supplemented with ampicillin (50 μg/ml) or chloramphenicol (30 μg/ml) as needed. E. coli strains were grown at 37°C except for complementation tests with temperature-sensitive strains, which were grown at 32°C (permissive) or 42°C (nonpermissive) on LB agar containing the appropriate antibiotic and no added NaCl. Overnight growth of colonies or several hours of growth in broth were usually sufficient to detect fluorescence from fusions with the improved GFP mutant.
Plasmid Constructions.
Plasmid pHS16 (from Henrik Stotz, Stanford University) contains GFP cloned on a BamHI fragment, with HindIII, XbaI, and XhoI sites immediately upstream of the ATG start codon. To facilitate cloning and expression of GFP and GFP fusion proteins, the 0.8-kb BamHI fragment from pHS16 was cloned into the BamHI site of pBC SK+ (Stratagene) such that GFP was fused to the lac promoter and the N terminus of lacZ. This plasmid, pCSK100, was used for the C-terminal GFP fusions to E. coli ftsZ and ftsA (pZG and pAG). E. coli ftsZ and ftsA lacking stop codons were amplified by PCR as described (6) from plasmid pZAQ (26) containing E. coli ftsQAZ (obtained from D. RayChaudhuri, Tufts University School of Medicine). Primers for ftsZ included its native ribosome binding site linked to a SacI site and its penultimate codon linked to XbaI. Primers for ftsA also were designed with SacI and XbaI sites, except that the first residue of ftsA was fused to the first 20 amino acids of the vector lacZ. PCR products of 1.3 and 1.2 kb for ftsA and ftsZ, respectively, were cloned into SacI/XbaI-cleaved pCSK100, fusing the genes in-frame with GFP with 4 additional amino acids (SRLE) between them. Subsequently, the faster-folding more highly fluorescent M2 GFP mutant (38) (obtained from R. Valdivia, Stanford University) was substituted for the wild-type GFP in the fusions. The 73-kDa FtsA–GFP and 68-kDa FtsZ–GFP proteins were confirmed by immunoblot analysis with anti-GFP polyclonal antiserum (CLONTECH). The fusion junctions were confirmed by DNA sequencing.
Additional constructions were made as follows. To make the N-terminal GFP-FtsZ, the M2 GFP gene lacking a stop codon was amplified, cloned into the BamHI site of pBC SK+ in-frame with lacZ, and fused in-frame to PCR-amplified ftsZ cloned between the ClaI and ApaI sites downstream. The two deletions of FtsZ and C-terminal deletion of FtsA were made by PCR with the same SacI and XbaI cloning strategy. The Rhizobium meliloti FtsZ1 deletion fusion with GFP was made by inserting GFP into the unique NruI site of the ampicillin-resistant plasmid pJC06 (39). The FtsA N-terminal deletion was made by cleaving pAG with Ecl136II and BglII, filling-in the overhang, and religating. FtsA104, a Thr → Ala change at position 215, was made by mutagenic PCR. Deleted fusion proteins were confirmed by immunoblot analysis. Fusion plasmids used in this study are shown in Fig. 1. In addition, the compatible plasmid for overexpressing E. coli ftsZ was pKM4, which is ftsZ from pZAQ cloned into the tetracycline-resistant broad-host-range plasmid pWM176 under control of the tac promoter (40). Extra copies of lacIq were provided by cloning a lacIq cassette into the EcoRI site downstream of GFP.
Figure 1.
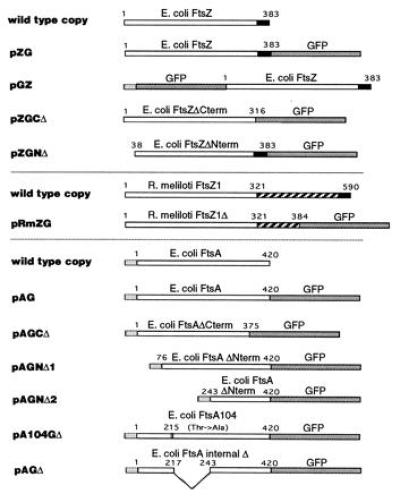
Plasmids expressing fusion proteins. Diagrammed below the wild-type E. coli FtsZ and FtsA and R. meliloti FtsZ1 protein diagrams are the various GFP fusions and deletions, all present on either pBluescript SK+ or pBC SK+. Numbers refer to the amino acid residue of FtsZ or FtsA at relevant junctions. Open boxes for FtsZ represent the conserved N terminus and for FtsA represent the entire encoded protein. Solid boxes denote the variable C terminus of FtsZ (residues 317–383 in E. coli FtsZ). Dark shaded boxes denote GFP and light shaded boxes denote the N-terminal 20 amino acids of vectorlacZ from the start codon to the SacI site. Hatched boxes represent the divergent and unusually long C terminus of R. meliloti FtsZ1.
Microscopic Techniques.
In general, cells were resuspended in LB and an equal volume of warm 2% low-melting-temperature agarose (Fisher) in LB, and 10 μl of the cells–agarose solution was immediately dropped on a glass slide and covered with a cover glass. For most images, Kodak TMAX 3200 print film or 400 Elite slide film was used to capture fluorescence images obtained with a Zeiss Axioskop [Zeiss ×100 oil-immersion objective, 100-W HBO lamp, standard fluorescein isothiocyanate filter set for GFP and a 4′,6-diamidino-2-phenylindole (DAPI) filter set for DNA]. DAPI-GFP colocalization in normal-length cells was captured digitally with the Image1 system (Universal Imaging, West Chester, PA) using a silicon-intensified target camera and a Nikon Diaphot microscope equipped with a 75-W XBO lamp and a Nikon ×100 oil-immersion objective. Three-dimensional reconstruction of cell fluorescence was performed with a DeltaVision wide-field optical sectioning microscope (Applied Precision, Mercer Island, WA) and visualized with a chilled charge-coupled device camera. Twelve to 16 optical sections were acquired every 0.1–0.2 μm on the x axis. Deconvolution of raw data was performed with 15 rounds of iteration and projections were made with the “brightest point” method. In all cases, images were compiled in Adobe photoshop and printed on a Tektronix dye-sublimation printer.
RESULTS
Characterization of FtsZ–GFP and FtsA–GFP Fusions.
To visualize FtsZ and FtsA proteins in living E. coli cells, merodiploids were constructed that contained GFP fusions to E. coli ftsA or ftsZ on multicopy plasmids (Fig. 1) in addition to the wild-type ftsZ and ftsA chromosomal loci. Expression of the fusions was under control of the lac promoter/operator on the plasmids and lacIq either on the same plasmid, a compatible plasmid, or on an F′. The GFP fusion to the C terminus of ftsZ contained the native ftsZ ribosome binding site, but the N-terminal ftsZ fusion and the ftsA-GFP C-terminal fusion used the lacZ ribosome binding site and the 5′ end of lacZ. Using anti-FtsZ antibodies, we found that the amount of uninduced FtsZ–GFP from pZG was usually roughly equal to or less than levels of wild-type FtsZ (data not shown). On the other hand, immunoblot experiments with anti-GFP antibody suggested that the level of FtsA–GFP synthesized from pAG in uninduced cells was much greater than the normal 50–200 molecules of wild-type FtsA. FtsA–GFP was brought closer to wild-type levels by introducing additional copies of lacIq, with the result that FtsA–GFP was barely detectable by immunoblot analysis unless expression was increased with isopropyl β-d-thiogalactoside (IPTG).
The effects of the fusions on cell physiology were examined. Cells with FtsZ–GFP and FtsA–GFP fusions containing additional copies of lacIq, either on a separate plasmid or cloned directly into pAG and pZG, were usually nonfilamentous. When expression was induced with increasing amounts of IPTG, cell division was increasingly inhibited and cells became more filamentous (data not shown). Since cell division inhibition also occurs when wild-type FtsZ and FtsA are overexpressed, it was not clear how much of this phenotype was due to the GFP tag. To test whether the fusion proteins were sufficient for cell division in the absence of wild-type FtsZ or FtsA, several different plasmids expressing the fusions as well as wild-type ftsA and ftsZ (on pZAQ) were introduced into E. coli strains containing temperature-sensitive mutations ftsZ84 or ftsA1882 and lacIq. Whereas strains containing pZAQ or a pBluescript derivative containing ftsZ under lac promoter control were able to form colonies at the restrictive temperature, those with only pAG or pZG could not, suggesting that FtsZ–GFP and FtsA–GFP are not fully competent for septation. It is unlikely that the lack of complementation for FtsZ–GFP, at least, was due to inappropriate fusion protein expression levels, since the complementing Plac-FtsZ plasmid has the same regulation and copy number. Attempts to obtain complementing fusions by increasing or decreasing transcription from the lac promoter, making an N-terminal fusion (pGZ) or by inserting a peptide linker between FtsZ and GFP in pZG were unsuccessful.
FtsZ–GFP Localizes to Rings at E. coli Division Sites.
The localization and structure of FtsZ–GFP were characterized in living cells taken from freshly grown colonies or from mid- to late logarithmic liquid cultures. The general localization patterns were similar under different growth conditions, including growth at lower temperatures or in minimal medium and in different host strains (data not shown). As long as the level of FtsZ–GFP in pZG/JM109 was equivalent to or lower than that of wild-type FtsZ, a high percentage of normal-length cells expressing the fusion exhibited a fluorescent band or double band at the cell midpoint (Fig. 2A). These bands were detectable only with fluorescence and not by phase-contrast microscopy, indicating that they were true cytoskeletal structures and not insoluble inclusions. Bands were observed in cells showing no signs of cytokinetic constriction, and bands or compact foci were also observed at constrictions representing intermediate to late stages of division (Fig. 2 B and C). Moreover, microscopic time-lapse studies of individual cells showed that cells containing fluorescence at the constriction could fully divide (Q. Sun and W.M., unpublished results), demonstrating that FtsZ–GFP can copolymerize with the FtsZ ring and allow normal septation. This result suggests that the lack of complementation may be due to the inability of FtsZ–GFP to localize or polymerize properly on its own. Double bands could either represent two adjacent FtsZ rings or a short FtsZ spiral structure. It is unclear why some cells exhibited double bands and others did not, although there may be a correlation with the size of the internucleoid space (see below). Control cells synthesizing only GFP in the same expression system were always uniformly fluorescent (Fig. 2D). Cells containing pGZ, the N-terminal fusion of FtsZ to GFP, also showed midcell localization (data not shown).
Figure 2.
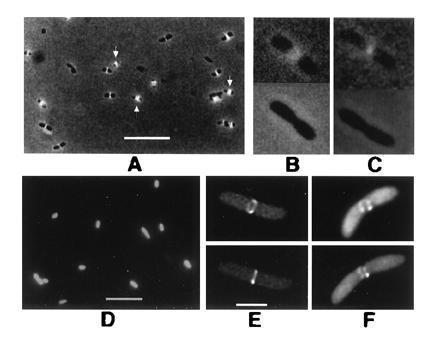
FtsZ–GFP forms rings at midpoints of E. coli cells. Cells in A–D were viewed by conventional fluorescence; cells in E and F were imaged by deconvolution. All were from colonies on plates without IPTG. (A) JM109/pZG, showing localization to cell midpoints. Arrows point to double bands. (B and C) FtsZ–GFP fluorescence (JM109/pZG) in cells undergoing visible septation. (Upper) With fluorescence/phase-contrast. (Lower) Phase-contrast only. (D) JM109 containing pCSK100 (GFP only). (E) JM109/pZG cell with FtsZ–GFP ring. Upper and Lower are two different viewing angles of the same cell. (F) Same as E except with incomplete fluorescent ring. (Bars: A and D, 5 μm; B, C, E, and F, 1 μm; bar shown in E only.)
To confirm that the midcell bands were rings, living JM109 cells containing FtsZ–GFP (pZG) in late logarithmic growth were imaged in three dimensions using deconvolution microscopy. Many cells contained midcell rings with an obvious lumen (Fig. 2E). These rings were also observed by conventional fluorescence microscopy but were difficult to photograph. Cells with incomplete rings were also occasionally observed (Fig. 2F); although these structures could be an artifact of the GFP tag, they coincide with midcell invagination and could represent a genuine intermediate in ring formation.
To determine whether the midcell rings localized between the two segregated daughter chromosomes, living cells were stained with DAPI and the positions of both blue (DAPI) and green (GFP) were observed. Green fluorescent bands predominantly localized between nucleoids (Fig. 3A). Some of these cells had no visible constrictions, suggesting that the images captured cells between DNA segregation and septation initiation (data not shown). Interestingly, when FtsZ–GFP double bands were observed, they were accompanied by a larger internucleoid space (Fig. 3B). One possible explanation for this result is that FtsZ rings only assemble in regions lacking DNA, and if the region is large enough, two adjacent rings or a tight spiral could form. Alternatively, duplicated periseptal annuli may be present in some cells (41), defining two sites for ring formation.
Figure 3.
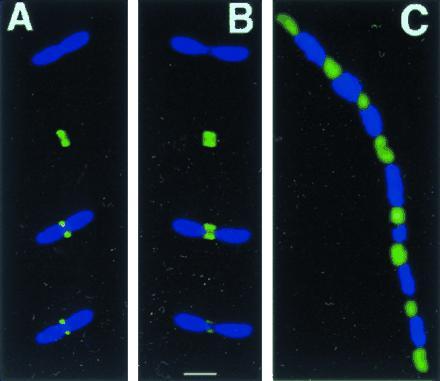
FtsZ–GFP localizes to internucleoid regions. (A and B) Individual cells of JM109/pZG stained with DAPI (1 μg/ml), viewed for DAPI fluorescence only (top part), GFP fluorescence only (second part from top), and DAPI + GFP (lower two parts, composite images, with lowest part darkened to improve visualization of the internucleoid space). (C) Filamentous cell (JM109/pZG) in a population of mostly normal cells, imaged for both DAPI and GFP. (Bar = 1 μm.)
These results confirm that the FtsZ rings seen in cross-sectional immunogold-stained cells are present in living E. coli cells and show that FtsZ and nucleoid positioning may be mutually exclusive. Despite the inability of the FtsZ–GFP fusion to fully complement an ftsZ mutant, it appears to localize and polymerize with wild-type FtsZ, acting as a sensitive and accurate tracer.
Localized Aggregates and Spiral Tubular Polymers with FtsZ–GFP in Vivo.
Although rings and incomplete rings were consistently observed when FtsZ–GFP was at levels lower than wild-type FtsZ, higher levels of FtsZ–GFP expressed from pZG or pGZ correlated with the appearance of other structures such as aggregates (dots) and spiral tubules. Such higher expression levels could be observed in some JM105 or JM109 cells containing just a single copy of lacIq and almost always after inducing with IPTG. Cells containing these unusual structures were usually filamentous, suggesting that the structures inhibited septation.
There were three general classes of fluorescence patterns. The first was a series of regularly spaced bands, probably rings, that extended the entire length of the filamentous cells. Their regular spacing of about 3 μm suggested that they were located at potential division sites (Fig. 4A). These structures may represent multiple FtsZ rings that were assembled but unable to continue the septation process. The second class of fluorescence pattern consisted of regularly spaced dots (Fig. 4B) that were not inclusion bodies since they were not visible with phase-contrast optics alone. The spacing of the dots was remarkably uniform in any given cell, but ranged anywhere between 1 μm and as much as 5 μm, depending on the cell. The dots were located consistently between nucleoids, strongly suggesting that this form of FtsZ–GFP was localizing to potential division sites (Fig. 3C). The third class of fluorescence pattern consisted of spiral tubules (Fig. 4 C–E). The spirals were observed in both compressed and elongated forms (data not shown) and could be observed in nonfilamentous cells (Fig. 4 C and D) as well as filamentous cells (Fig. 4E).
Figure 4.
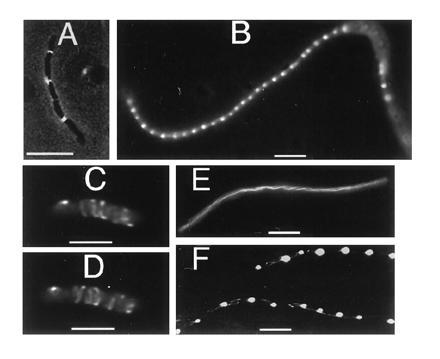
Tubules, bands, and dots in cells with FtsZ–GFP. JM109/pZG cells were from colonies on plates without IPTG. (A) Moderately filamentous cell showing regularly spaced bands. (B) Filamentous cell in a population of mostly normal cells, showing regularly spaced dots. (C) Nonfilamentous cell containing spiral polymers, imaged with deconvolution. (D) Same cell as in C but from a different angle. (E) Filamentous cell with spiral polymers. (F) Filamentous cells showing both spiral polymers and regularly spaced dots. (Bars: A, B, E, and F, 5 μm; C and D, 1 μm.)
These results suggest either that the tubules and dots are artifacts caused by the GFP tag or that wild-type FtsZ itself forms tubules and aggregates in vivo under certain conditions. Our data for FtsA–GFP suggest that at least the spirals are formed by wild-type FtsZ (see below). The observed tubules and dot-like aggregates in the same population and even within individual cells (Fig. 4F) suggest that FtsZ in aggregated or sequestered form may be able to change to a tubular polymerized form while still maintaining its association with spatial markers on the cell wall.
FtsA–GFP Colocalizes with FtsZ.
The localization of FtsA–GFP fusions in E. coli was visualized using similar techniques. When expression was low in the presence of multiple copies of lacIq, FtsA–GFP expressed from pAG consistently localized to midcell or to potential division sites (Fig. 5 A and B). FtsA–GFP also was associated with a midcell ring-like structure similar to that of FtsZ–GFP (Fig. 5 C and D). The rings appeared distorted, perhaps due to a uneven sparse distribution of FtsA–GFP molecules binding to the FtsZ ring or an effect on FtsZ ring structure itself. As with cells containing FtsZ–GFP, some FtsA–GFP cells from stationary-phase colonies contained visible double bands that probably represent localization to double FtsZ rings (Fig. 5A). As with FtsZ–GFP, FtsA–GFP fluorescence was sometimes observed at the division site during later stages of septation. When FtsA–GFP was overproduced by derepressing the lac promoter, such that cellular levels of FtsA–GFP were higher than that of FtsZ–GFP, the cells were filamentous and contained bulges characteristic of FtsA overproduction (28). FtsA–GFP fluorescence was dispersed throughout the cells, although diffuse areas of localization could be seen at regular intervals down the filament and within the bulges (Fig. 5F).
Figure 5.
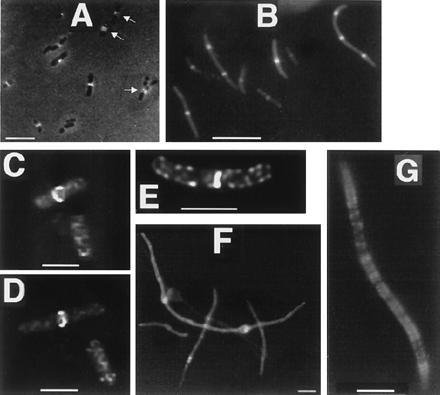
FtsA–GFP localization. (A–E) Cells with pAG and extra copies of lacIq, from colonies on plates without IPTG. (A) JM109/pAG cells with FtsA–GFP fluorescence localized to cell midpoints. Arrows point to double bands. (B) MC1061/pAG cells, with lacIq cloned into the plasmid. (C) Deconvolution image of JM109/pAG cells, one showing FtsA–GFP localization to a midcell ring, the other showing spiral structures. (D) Same cells as in C, at a different angle. (E) Deconvolution image of JM109/pAG cell showing midcell fluorescence and spiral structures. (F) Filamentous JM109/pAG cells with no extra copies of lacIq; higher levels of FtsA–GFP cause bulges and more diffuse fluorescence. (G) Filamentous JM105/pAG/pMK4 cell expressing FtsA–GFP and wild-type FtsZ, with IPTG. (Bars: A, B, F, and G, 5 μm; C and D, 1 μm.)
FtsA–GFP also was associated with nonring structures. (i) Fluorescence could often be seen at the cell surface in addition to the midcell site, as if a subpopulation of molecules were being sequestered at the membrane (data not shown). This is consistent with the evidence that the membrane localization of FtsA depends on its phosphorylation state (20). (ii) We sometimes observed nonfilamentous cells expressing FtsA–GFP that contained short fluorescent spiral structures equivalent to those observed with FtsZ–GFP (Fig. 5 E, also bottom cell in C and D). To test whether these spirals represented wild-type FtsZ, we overproduced FtsZ in the presence of FtsA–GFP by IPTG induction of a JM105 strain containing both pAG and pKM4. We observed filamentous cells that contained fluorescent spirals (Fig. 5G) similar to those seen with FtsZ–GFP in filamentous cells (Fig. 4E).
These observations have several important implications. First, FtsA itself is unlikely to form polymers because of its low abundance, so it is most likely that fluorescent rings, double rings, or spirals observed with FtsA–GFP are an indirect effect of FtsA–GFP decorating FtsZ structures. The increased number of spirals decorated by FtsA–GFP at increased FtsZ concentrations supports the idea that wild-type FtsZ itself can switch into a spiral form when at higher concentrations; generation of such nonproductive structures could explain how a large excess of FtsZ inhibits cell division.
Function of FtsZ Domains.
To address the domain structure and function of FtsA and FtsZ, we used the GFP tag to determine the in vivo localization and structure of several deletions of FtsA and FtsZ shown in Fig. 1. To test the possible role of the divergent C terminus of FtsZ, a 67-amino acid C-terminal domain was deleted from FtsZ, leaving the N-terminal 316 amino acids attached to GFP (pZGCΔ). This domain was chosen because an abrupt change from conserved to nonconserved residues occurs at position 316 with only a small group of conserved residues at the extreme C terminus. Cells with pZGCΔ exhibited both spiral polymers similar to those in Fig. 4E and very thick polymer sheets (Fig. 6A) reminiscent of protofilament sheets seen with purified FtsZ. Cells expressing this fusion were highly filamentous, even though the level of protein by immunoblot was no higher than that of the full-length fusion (data not shown). This suggests that the truncated fusion is a more potent division inhibitor than the full-length fusion. The fluorescence was also significantly brighter than that for the full-length fusion, perhaps because the polymers are unusually stable. Occasional ring-like bands with very weak fluorescence were found that may represent FtsZΔ–GFP mixing with wild-type FtsZ. Therefore, the the divergent C-terminal 67 residues of FtsZ are not required for polymerization but may have a role in regulating polymer stability and morphology.
Figure 6.
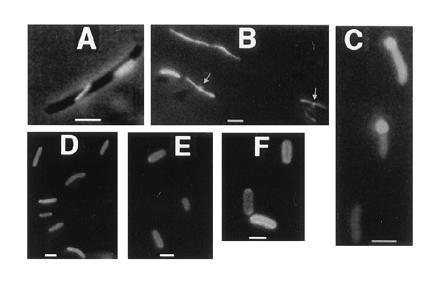
Effects of deletions of GFP-tagged FtsA and FtsZ. (A) JM109/pZGCΔ cell showing typical large unlocalized structures. (B) JM109/pRmZG cells with axial polymers produced by R. meliloti FtsZ1Δ-GFP. Arrows point to midcell localization observed in a subset of cells. (C) JM105/pZGNΔ cells showing uniform fluorescence; the regions of bright fluorescence at the poles seen in some cells are large inclusion bodies. (D) JM105/pAGCΔ. (E) JM105/pAGNΔ1. (F) JM105/pAGNΔ2 cells; cell surface fluorescence is especially apparent. (Bars = 1 μm.)
When the conserved N-terminal domain of R. meliloti FtsZ1 corresponding to E. coli FtsZ residues 1–316 was fused to GFP (pRmZG) and expressed in E. coli, virtually all cells contained a single straight fluorescent tubule extending from pole to pole, and some cells showed midcell fluorescence of lower magnitude (Fig. 6B). This result suggests that the R. meliloti fusion, like the truncated E. coli fusion, can both polymerize on its own in a different host and copolymerize with wild-type E. coli FtsZ at the midcell ring. We can conclude that the C terminus of R. meliloti FtsZ1 is also not required for polymerization. Overexpression of the R. meliloti fusion causes lethal filamentation of E. coli cells, suggesting that it may interact with E. coli FtsZ nonproductively by copolymerization.
With pZGNΔ, expressing an N-terminal deletion of 37 residues of FtsZ in pGZ, no polymerization or localization was detected (Fig. 6C). Also, despite formation of polar inclusion bodies, which are often seen with overexpressed proteins, overexpression of this fusion had no significant detrimental effect on cell division. This is probably because copolymerization is required for cell division inhibition during FtsZ overexpression. Therefore, this region, which does not include the GTP binding box but does include several highly conserved residues, appears to be essential for FtsZ polymerization.
Search for FtsA Localization Domains.
As a first step to identify domains in FtsA important for localization to the FtsZ ring, we constructed a series of deletions of pAG. The first deletion (pAGCΔ) removed the C-terminal 45 amino acids, leaving the conserved ATP binding motif IVLTGG intact but removing the “connect 2” domain (21). In addition, two N-terminal deletions were made that removed the first 75 amino acids (pAGNΔ1) and 242 amino acids (pAGNΔ2), respectively, and an internal in-frame deletion was made that removed residues 211–242 (pAGΔ). These fusions were expressed in JM105 along with pAG as a positive control and examined for localization. Interestingly, all four deleted fusion proteins were able to associate with the cell surface, but none localized to midcell in hundreds of cells examined for each under several different expression conditions (Fig. 6 D–F). Cells expressing the deleted protein fusions were all of normal length, suggesting that the fusions are nonfunctional. In contrast, a GFP fusion to FtsA104, which is defective in phosphorylation and ATP binding but still can complement ftsA mutants (20), could localize to the FtsZ ring (data not shown). This supports the idea that phosphorylation and ATP binding are not essential for FtsA localization. The apparent ability of all four deleted fusion proteins to associate with the cell surface suggests that a domain sufficient for membrane partitioning of FtsA lies between residues 243 and 375. Our results suggest that several segments of FtsA may contribute to a localization domain defined by the tertiary structure.
DISCUSSION
In a first step toward a whole-cell assay system for studying septation protein targeting and structure, we have detected localized structures for FtsZ–GFP and FtsA–GFP in living E. coli cells. The FtsZ–GFP rings confirm published data with fixed cells and demonstrate the power and sensitivity of the technique. The FtsA–GFP localization to rings is, to our knowledge, the first demonstration of targeting of a protein other than FtsZ to the E. coli division site and supports the idea of a septation protein complex.
We have also identified other unprecedented structures such as spiral tubules, properly localized aggregates, and incomplete rings. Our data suggest that FtsZ switches to a spiral in vivo when its concentration is higher than normal. Although the dots and incomplete rings may be artifacts influenced by the GFP tag, they also may reflect blocked intermediates in the normal dynamics of FtsZ in the cell. For example, FtsZ could initially assemble a small structure at one position at midcell by analogy to γ-tubulin, and then nucleate formation of tubules from that site. Dots could represent correctly localized FtsZ unable to form tubules, perhaps because of intercalation of FtsZ–GFP, and incomplete rings could be partially assembled tubules that became blocked at a later stage.
The most likely reason why FtsZ–GFP polymerizes, localizes, and yet fails to complement the ftsZ84 mutant is that FtsZ–GFP cannot localize on its own but can copolymerize with wild-type FtsZ. Once the ratio of FtsZ–GFP to FtsZ reaches a certain threshold, FtsZ–GFP might no longer be able to act as a benign tracer and instead could perturb FtsZ polymer function, perhaps by preventing proper localization. The potential reasons behind the inability of FtsA–GFP to complement the ftsA mutant are less obvious. FtsA–GFP does not seem to inhibit septation when repressed by multiple copies of lacIq, since rapidly growing cells exhibiting midcell localization are not filamentous. In addition, FtsA–GFP often localizes to invaginating septa, retains wild-type activity by binding to FtsZ polymers, and causes an FtsA-specific phenotype when overexpressed. Further study of the structure and multiple functions of FtsA and FtsZ should shed light on the specific functions missing or abnormal in FtsA–GFP and FtsZ–GFP.
Except for the full-length version and FtsA104, the various segments of FtsA tagged with GFP reported herein all failed to show midcell localization. This suggests that proper tertiary structure may be essential for FtsA localization to midcell and that no single midcell localization domain may exist in the primary sequence. Expression of these deleted FtsA–GFP proteins, in contrast to the full-length fusion on pAG, did not significantly affect cell division. This may be due to their apparent failure to interact with FtsZ, suggesting that division inhibition due to excess FtsA may require the presence of FtsA at midcell.
We have provided further evidence for the structural similarity between FtsZ and tubulin. (i) FtsZ–GFP, even from another organism, can form long tubules in E. coli cells. (ii) The variable C termini of E. coli and R. meliloti FtsZ are not essential for polymerization, and neither is the variable C terminus of tubulin (42). As with tubulin C-terminal truncations, polymers made by the C-terminal-deleted derivative of E. coli FtsZ are unusually large and stable. This is perhaps analogous to the essential role of the C terminus of tubulin in dynamic instability (42). The lack of complementation by the GFP fusion may in part be due to the effect of GFP on the integrity of the adjacent FtsZ C terminus, since the C terminus may be essential for function (43). We speculate that the essential role of the C terminus is to regulate a switch between midcell rings and other apparently nonproductive structures. Further studies on the structure of FtsZ may provide further support for its similarity to tubulin.
The data presented herein argue that the GFP tagging approach will be a valuable complement to studies of mutant phenotypes and protein–protein interactions with purified components. Localization of these and other GFP fusions in cell division mutants should allow a more detailed understanding of the steps in the septation pathway. In addition, GFP should allow dynamic tracking of protein movement and turnover in cells, which should help elucidate the mechanisms of prokaryotic cell division from a whole-cell perspective.
Acknowledgments
We are indebted to T. Vida for valuable advice and the use of his fluorescence microscope facility. We thank P. Christie, G. Singh, D. Neal, and M. Kumar for help with cloning GFP; the University of Texas, Department of Pharmacology for use of their graphics facility; H. Stotz, R. Tsien, R. Valdivia, and B. Cormack for GFP clones; J. Lutkenhaus and D. RayChaudhuri for ftsZ strains and plasmids, and H. Kaplan and A. Dombroski for comments on the manuscript. We thank the Howard Hughes Medical Institute for their support of this work. In addition, this work was supported by National Science Foundation Grants MCB-9410840 and MCB-9513521 (to W.M.).
Footnotes
Abbreviations: GFP, green fluorescent protein; DAPI, 4′,6-diamidino-2-phenylindole; IPTG, isopropyl β-d-thiogalactoside.
References
- 1.Rothfield L I, Zhao C-R. Cell. 1996;84:183–186. doi: 10.1016/s0092-8674(00)80971-x. [DOI] [PubMed] [Google Scholar]
- 2.Chang F, Nurse P. Cell. 1996;84:191–194. doi: 10.1016/s0092-8674(00)80973-3. [DOI] [PubMed] [Google Scholar]
- 3.Donachie W D. Annu Rev Microbiol. 1993;47:199–230. doi: 10.1146/annurev.mi.47.100193.001215. [DOI] [PubMed] [Google Scholar]
- 4.Lutkenhaus J. Mol Microbiol. 1993;9:403–410. doi: 10.1111/j.1365-2958.1993.tb01701.x. [DOI] [PubMed] [Google Scholar]
- 5.Corton J C, Ward J E, Lutkenhaus J. J Bacteriol. 1987;169:1–7. doi: 10.1128/jb.169.1.1-7.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Margolin W, Wang R, Kumar M. J Bacteriol. 1996;178:1320–1327. doi: 10.1128/jb.178.5.1320-1327.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osteryoung K W, Vierling E. Nature (London) 1995;376:473–474. doi: 10.1038/376473b0. [DOI] [PubMed] [Google Scholar]
- 8.Dai K, Lutkenhaus J. J Bacteriol. 1992;174:6145–6151. doi: 10.1128/jb.174.19.6145-6151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Lutkenhaus J. Mol Microbiol. 1993;9:435–442. doi: 10.1111/j.1365-2958.1993.tb01705.x. [DOI] [PubMed] [Google Scholar]
- 10.Bi E, Lutkenhaus J. Nature (London) 1991;354:161–164. doi: 10.1038/354161a0. [DOI] [PubMed] [Google Scholar]
- 11.Chant J. Cell. 1996;84:187–190. doi: 10.1016/s0092-8674(00)80972-1. [DOI] [PubMed] [Google Scholar]
- 12.Traas J, Bellini C, Nacry P, Kronenberger J, Bouchez D, Caboche M. Nature (London) 1995;375:676–677. [Google Scholar]
- 13.Mukherjee A, Lutkenhaus J. J Bacteriol. 1994;176:2754–2758. doi: 10.1128/jb.176.9.2754-2758.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mukherjee A, Dai K, Lutkenhaus J. Proc Natl Acad Sci USA. 1993;90:1053–1057. doi: 10.1073/pnas.90.3.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.RayChaudhuri D, Park J T. Nature (London) 1992;359:251–254. doi: 10.1038/359251a0. [DOI] [PubMed] [Google Scholar]
- 16.de Boer P, Crossley R, Rothfield L. Nature (London) 1992;359:254–256. doi: 10.1038/359254a0. [DOI] [PubMed] [Google Scholar]
- 17.Bramhill D, Thompson C M. Proc Natl Acad Sci USA. 1994;91:5813–5817. doi: 10.1073/pnas.91.13.5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erickson H P, Taylor D W, Taylor K A, Bramhill D. Proc Natl Acad Sci USA. 1996;93:519–523. doi: 10.1073/pnas.93.1.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vicente M, Errington J. Mol Microbiol. 1996;20:1–7. doi: 10.1111/j.1365-2958.1996.tb02482.x. [DOI] [PubMed] [Google Scholar]
- 20.Sanchez M, Valencia A, Ferrandiz M-J, Sandler C, Vicente M. EMBO J. 1994;13:4919–4925. doi: 10.1002/j.1460-2075.1994.tb06819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bork P, Sander C, Valencia A. Proc Natl Acad Sci USA. 1992;89:7290–7294. doi: 10.1073/pnas.89.16.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pla J, Dopazo A, Vicente M. J Bacteriol. 1990;172:5097–5102. doi: 10.1128/jb.172.9.5097-5102.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tormo A, Martinez-Salas E, Vicente M. J Bacteriol. 1980;141:806–813. doi: 10.1128/jb.141.2.806-813.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Addinall S G, Bi E, Lutkenhaus J. J Bacteriol. 1996;178:3877–3884. doi: 10.1128/jb.178.13.3877-3884.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Gayda R C. Mol Microbiol. 1992;6:2517–2524. doi: 10.1111/j.1365-2958.1992.tb01428.x. [DOI] [PubMed] [Google Scholar]
- 26.Ward J E, Lutkenhaus J. Cell. 1985;42:941–949. doi: 10.1016/0092-8674(85)90290-9. [DOI] [PubMed] [Google Scholar]
- 27.Dewar S J, Begg K J, Donachie W D. J Bacteriol. 1992;174:6314–6316. doi: 10.1128/jb.174.19.6314-6316.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H, Henk M C, Gayda R C. Curr Microbiol. 1993;26:175–181. [Google Scholar]
- 29.Wang H, Gayda R C. J Bacteriol. 1990;172:4736–4740. doi: 10.1128/jb.172.8.4736-4740.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prasher D C. Trends Genet. 1995;11:320–323. doi: 10.1016/s0168-9525(00)89090-3. [DOI] [PubMed] [Google Scholar]
- 31.Stearns T. Curr Biol. 1995;5:262–264. doi: 10.1016/s0960-9822(95)00056-x. [DOI] [PubMed] [Google Scholar]
- 32.Heim R, Prasher D C, Tsien R Y. Proc Natl Acad Sci USA. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chalfie M, Tu Y, Euskirchen G, Ward W, Prasher D C. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 34.Arigoni F, Pogliano K, Webb C D, Stragier P, Losick R. Science. 1995;270:637–640. doi: 10.1126/science.270.5236.637. [DOI] [PubMed] [Google Scholar]
- 35.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 36.Hirota Y, Ryter A, Jacob F. Cold Spring Harbor Symp Quant Biol. 1968;33:677–694. doi: 10.1101/sqb.1968.033.01.077. [DOI] [PubMed] [Google Scholar]
- 37.Walker J R, Kovarik A, Allan J, Gustafson R A. J Bacteriol. 1975;123:693–703. doi: 10.1128/jb.123.2.693-703.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cormack B P, Valdivia R H, Falkow S. Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 39.Margolin W, Corbo J C, Long S R. J Bacteriol. 1991;173:5822–5830. doi: 10.1128/jb.173.18.5822-5830.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Margolin W, Long S R. J Bacteriol. 1994;176:2033–2043. doi: 10.1128/jb.176.7.2033-2043.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MacAlister T J, MacDonald B, Rothfield L I. Proc Natl Acad Sci USA. 1983;80:1372–1376. doi: 10.1073/pnas.80.5.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sackett D L, Bhattacharyya B, Wolff J. J Biol Chem. 1985;260:43–45. [PubMed] [Google Scholar]
- 43.Wang X, Lutkenhaus J. J Bacteriol. 1996;178:2314–2319. doi: 10.1128/jb.178.8.2314-2319.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]