

Abstract
Effective hematopoiesis requires the commitment of pluripotent and multipotent stem cells to distinct differentiation pathways, proliferation and maturation of cells in the various lineages, and preservation of pluripotent progenitors to provide continuous renewal of mature blood cells. While the importance of positive and negative cytokines in regulating proliferation and maturation of hematopoietic cells has been well documented, the factors and molecular processes involved in lineage commitment and self-renewal of multipotent progenitors have not yet been defined. In other developmental systems, cellular interactions mediated by members of the Notch gene family have been shown to influence cell fate determination by multipotent progenitors. We previously described the expression of the human Notch1 homolog, TAN-1, in immature hematopoietic precursors. We now demonstrate that constitutive expression of the activated intracellular domain of mouse Notch1 in 32D myeloid progenitors inhibits granulocytic differentiation and permits expansion of undifferentiated cells, findings consistent with the known function of Notch in other systems.
Keywords: Notch1 gene, hematopoiesis, 32D cells
Members of the Notch gene family encode large transmembrane proteins that play central roles in mediating cell fate decisions during early development in a wide variety of different tissues and organisms (for review, see refs. 1–5). The high degree of structural conservation among family members (6–12), as well as genetic and molecular studies in several systems (1–3, 13–15), provide considerable evidence for evolutionary conservation of Notch function from invertebrate species to mammals. The protein products of Notch family members consist of the following: an extracellular domain containing tandem epidermal growth factor repeats and three lin-12/Notch repeats; a single transmembrane domain; and an intracellular domain that contains six cdc10/SWI6/ankyrin repeats, putative nuclear localization signals, and a C-terminal OPA/PEST region (Fig. 1). Cellular interactions mediated by the extracellular domain binding to its ligand(s) are thought to activate the intracellular domain, resulting in inhibition of differentiation along a particular pathway, but leaving cells competent to adopt alternative fates (2, 4, 5).
Figure 1.
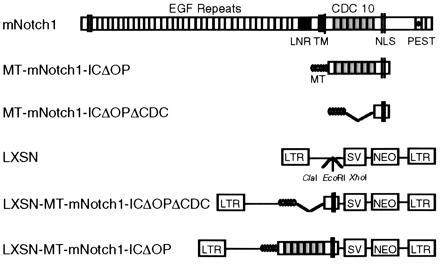
Diagram of the mNotch1 gene product and retroviral constructs. The molecule consists of an extracellular domain containing 36 tandem epidermal growth factor (EGF) repeats and 3 lin-12/Notch (LNR) repeats; a single transmembrane domain (TM); and an intracellular domain containing 6 cdc10/SWI6/ankyrin repeats, putative nuclear localization signals (NLS) and a C-terminal OPA/PEST region. The activated mNotch1 construct (MT-mNotch-ICΔOP) consists of the region of the intracellular domain including the cdc10 repeats and 3′ nuclear localization signals. The constructs included 5′ myc epitope tags (MT) to facilitate detection of protein expression. The control construct MT-mNotch1-ICΔOPΔCDC lacks the 5′ end of mNotch through the cdc10 repeats, but otherwise corresponds to MT-mNotch1-ICΔOP. Constructs were cloned into the LXSN retroviral vector that had been modified to contain a ClaI restriction site.
Studies of Notch in various systems have demonstrated that unregulated expression of the intracellular domain results in an “activated ” Notch phenotype and that the cdc10 repeat region is crucial for intracellular signal transduction (13–20). In these studies, constitutive expression of truncated Notch molecules results in phenotypic effects consistent with those observed or expected from activation of the full-length molecule by its extracellular ligand(s). These truncated Notch molecules lack most or all of the extracellular domain, contain only the intracellular domain, or contain only portions of the intracellular domain that include the cdc10 repeat region. Two mammalian Notch homologs, TAN-1 (hNotch1) and int-3, are similarly truncated in malignant cells (12, 21, 22). Taken together, these observations support the view that unregulated activity of intracellular Notch inhibits differentiation along a specific pathway, leaving undifferentiated cells capable of expanding or adopting a default pathway.
We previously described the expression of hNotch1 (TAN-1) in human bone marrow hematopoietic precursors (23). In those studies, we found expression of hNotch1 in CD34+ marrow cells, including both the immature subset that lacks expression of lineage-associated antigens (CD34+lin−) and the somewhat more mature lineage-positive (CD34+lin+) population. We hypothesized that Notch homologs influence cell fate determination at multiple steps during hematopoiesis, analogous to the role of Notch in other developmental systems. To explore the potential role of Notch activity in granulocytic differentiation, we have used retroviral transduction to express the active region of the intracellular domain of mNotch1 in the myeloid progenitor cell line, 32D. We find that constitutive expression of this activated form of mNotch1 in 32D cells inhibits granulocytic differentiation in response to granulocyte colony-stimulating factor (G-CSF), but leaves proliferative capacity intact. The findings presented here are consistent with the known role of Notch in other systems and, together with the expression of Notch in normal hematopoietic progenitors, suggest that Notch may play a similar role as a mediator of cell fate specification in hematopoietic cells.
MATERIALS AND METHODS
Preparation of Retroviral Vectors Containing mNotch Constructs.
The retroviral vector containing the active region of the intracellular domain of mNotch1 was derived from a full-length mNotch1 clone (11) as follows: the intracellular domain of mNotch1 was cloned into the StuI site of the pCS2+6MT vector so that the coding sequence of mNotch1-IC (amino acid 1753) was in frame with the six myc epitope tags in the vector (15); the ClaI–XhoI fragment containing the MT plus the mNotch1 cdc10 repeats and nuclear localization signals (TKKFRFEE through KGCLLDSS) was then subcloned into a pLXSN retroviral vector that had been modified to contain a ClaI cloning site. The control construct mNotch1-ICΔOPΔCDC was derived from mNotch1-IC by deleting the 5′ end through the cdc10 repeats (RK); this fragment was cloned into pCS2+6MT and subcloned into pLXSN as for mNotch-ICΔOP. The resulting constructs included myc epitope tags (MT) to facilitate screening for protein expression using an anti-MT monoclonal antibody (9e10) (24). All constructs were verified by DNA sequencing. The intracellular domain of mNotch2 was PCR amplified from cDNA obtained from FDCP mixA4 cells, a murine factor-dependent multipotent hematopoietic cell line. PCR products obtained from several independent amplifications were gel purified, cloned into pGEM-T (Promega) and sequenced. The fragment of mNotch2 corresponding to the cdc10-nuclear localization signals region used for the mNotch1-ICΔOP construct (amino acids 1767–2156) predicts an amino acid sequence 98.7% identical to that of rat Notch2 (25). This fragment was subcloned into pCS2+6MT and the MT-mNotch2-ICΔOP fragment then cloned into the pLXSN vector.
Retroviral Transduction.
Retroviral producer cell lines were established using published protocols (26). Retroviral vectors were transfected by calcium phosphate precipitation into the ecotropic viral packaging cell line, PE501, and the transiently expressed virus was used to infect the amphotropic viral packaging cell line, PA317. Cells were selected in G418 and resistant clones were assayed by reverse transcription-PCR or Western blot for expression of the constructs. For transduction of 32D cells, PA317 producer cells were seeded on the bottom layer and 32D cells on the top layer of 24-mm transwells with a 3-μm pore. After 24 hr of cocultivation, polybrene (4 μg/ml) was added and medium (containing virus) was gently transferred from the bottom to the top layer of the transwell. Cells were incubated for an additional 24 hr. The 32D cells were then harvested, washed, and replated into 96-well plates in medium containing 1 mg/ml G418. Resistant clones for each construct were isolated, expanded, and evaluated by RT-PCR (LXSN retroviral vector alone) and/or Western blot (LXSN-MT-mNotch constructs) to confirm construct expression.
Western Blot Analysis.
Total cell lysates prepared from 5 × 105 cells of individual clones were electrophoresed through SDS/10% polyacrylamide gels, transferred to nitrocellulose membranes, and immunoblotted using the 9e10 (α-myc tag) monoclonal antibody. Immunostained proteins were detected using enhanced chemiluminescence Western blot reagents (Amersham). Films (BioMax; Kodak) were scanned and reproduced for publication using PhotoShop (Adobe Systems, Mountain View, CA) and canvas (Deneba, Miami, FL) software.
Cell Culture.
32D cells were maintained in Iscove’s modified Dulbecco’s medium with 10% fetal bovine serum and 10% Wehi-conditioned medium (WCM) as a source of interleukin 3 (IL-3). For differentiation experiments, 32D cell lines were harvested in log phase, washed, counted, and replated at constant density (2 × 105 cells per ml; 4 ml per well) in 6-well plates in Iscove’s modified Dulbecco’s medium containing 10% fetal bovine serum, 1% WCM, and 10 ng/ml recombinant human G-CSF (provided by Amgen). Differentiation experiments were done with all of the clones simultaneously, using the same lots of media and growth factors to avoid any variations in culture conditions. Viable cells were counted and Wright-stained cytospins were evaluated for granulocytic differentiation. Undifferentiated 32D cells generally have a single large, relatively round nucleus and scant dark blue cytoplasm containing few large granules. Criteria for granulocytic differentiation included nuclear segmentation, an increased cytoplasmic/nuclear ratio, and increased eosinophilia and granularity of the cytoplasm. Considerable care was taken to ensure consistent and unbiased evaluation of cellular morphology. All differential cell counts were done by the same individual (L.A.M.), repeated at least twice (100–200 cell differentials each time) on separate occasions, and cross-checked in random order to ensure consistency. Slides were also reviewed in a blinded fashion by several other individuals to verify the absence of observer bias.
RESULTS
To evaluate the effect of Notch activity on granulocytic differentiation, we used retroviral transduction to stably express the active region of the intracellular domain of mNotch1 in 32D myeloid progenitors. Murine 32D cells proliferate as undifferentiated blasts when maintained in IL-3, but differentiate into mature neutrophilic granulocytes when stimulated with G-CSF (27). We used a truncated mNotch molecule, comparable to activated Notch molecules known to be functional in other systems (14–16, 18, 20). This activated form of mNotch would be expected to result in phenotypic effects comparable to those resulting from activation of the intracellular domain though ligand binding. Fig. 1 shows the activated intracellular mNotch1 and control retroviral constructs. The active form, MT-mNotch1-ICΔOP, contains the cdc10 repeats and nuclear localization signals, but not the OPA/PEST sequences; control constructs consist of the pLXSN retrovirus alone and a construct derived from mNotch1-ICΔOP that lacks the cdc10 repeats (MT-mNotch1-ICΔOPΔCDC). After confirming construct expression, individual clones were evaluated for growth and differentiation characteristics in the presence and absence of differentiation signals (G-CSF).
32D cell clones were routinely maintained in media containing WCM as a source of IL-3. The day before the beginning of an experiment, cell cultures were split to constant density and fed with fresh media to ensure similar logarithmic-phase growth for all clones. On day 0, cells were washed and replated at constant density in media containing 10% WCM or 10 ng/ml G-CSF and 1% WCM. Cultures were evaluated daily for the total number of viable cells and the relative percentages of undifferentiated cells and mature granulocytes. Differential cell counts on Wright-stained cytospins were used to separate cells into three general categories: (i) undifferentiated (blasts), (ii) mature (bands and segmented granulocytes), and (iii) intermediate (myelocytes, metamyelocytes, and undetermined). The relative percentages and absolute numbers of undifferentiated and mature cells in cultures of mNotch1-ICΔOP-transduced cells compared with control clones and parental 32D cells were used to establish the effects of mNotch1 activation on granulocytic differentiation of 32D cells.
The Activated Intracellular Domain of mNotch1 Inhibits Granulocytic Differentiation of 32D Myeloid Progenitors.
As shown in Fig. 2, constitutive expression of activated mNotch1 inhibited G-CSF-induced differentiation of 32D cells. In this representative experiment, we evaluated six individual clones expressing the activated mNotch1-ICΔOP molecule, 10 control clones, and the parental cell line (32D WT) for growth and differentiation in response to G-CSF. When maintained in medium containing IL-3, but lacking G-CSF, 32D cells continued to proliferate as undifferentiated cells as expected (Fig. 2, top row). When stimulated with G-CSF, the 32D WT cells and the control lines (LXSN and mNotch1-ICΔOPΔCDC) showed a progressive decrease in the proportion of undifferentiated cells and a concomitant increase in the proportion of mature granulocytes (Fig. 2A). By day 6, <10% of the cells in these control cultures remained undifferentiated and an average of 35–45% had attained a mature granulocytic phenotype (Fig. 2B). In contrast, during the same period of time the mNotch1-ICΔOP lines never reached >10% mature granulocytes and the majority of cells (>55%) remained undifferentiated (Fig. 2 A and B Bottom).
Figure 2.
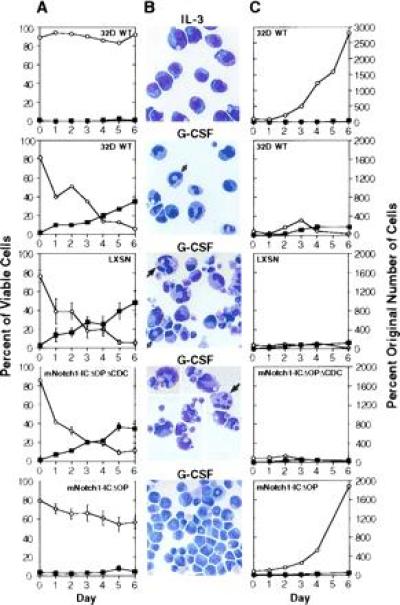
Effect of activated mNotch1 expression on the differentiation and proliferation of 32D cells. The parental 32D line (WT) and 32D cells transduced with the pLXSN retrovirus, pLXSN containing the active region of the intracellular domain of mNotch1 (mNotch1-ICΔOP), or a pLXSN-mNotch1 control lacking the cdc10 repeats (mNotch1-ICΔOPΔCDC) were evaluated for growth and differentiation in response to G-CSF. This figure represents one of four experiments, all having comparable results. (A) The relative percentages of viable cells remaining undifferentiated (○) or showing morphologic characteristics of mature granulocytes (▪) after successive days in culture. Plots for the retroviral lines represent the averages of four (LXSN) or six (mNotch1-ICΔOP and mNotch1-ICΔOPΔCDC) individual clones. Error bars = SEM for each set of clones; error bars are not included on the WT graphs since these represent a single population. (B) Representative Wright-stained cells. IL-3, undifferentiated parental 32D (WT) cells maintained in medium containing 10% WCM as a source of IL-3; G-CSF, 32D WT cells and representative clones expressing pLXSN, pLXSN-mNotch1-ICΔOPΔCDC or pLXSN-mNotch1-ICΔOP after 6 days in medium containing 1% WCM and 10 ng/ml G-CSF. The cells depicted in the second and fourth panels (32D WT and mNotch1-ICΔOPΔCDC) represent a single field condensed by deleting vacant areas to show a greater number of cells. The white and black arrows indicate band forms and segmented neutrophils, respectively. (C) The proliferation of undifferentiated cells (○) and mature granulocytes (▪) after successive days in culture with IL-3 (32D WT, Upper) or G-CSF. The total number of undifferentiated cells and mature granulocytes relative to the original number of cells plated are compared for the 32D parent line (WT) and each set of retroviral clones. Again the graphs represent the averages of four (LXSN) or six (mNotch1-ICΔOPΔCDC and mNotch1-ICΔOP) clones. SEMs were small; error bars are not depicted for the sake of clarity and would not change the impression created by the figure. Cells showing some characteristics of differentiation, but which were less mature than band cells were not included in either the undifferentiated or mature categories.
Expression of the Activated Intracellular Domain of mNotch1 Permits Expansion of Immature Myeloid Progenitors.
In addition to evaluating the relative proportions of undifferentiated and mature cells, we used the total number of viable cells and differential cell counts to determine the absolute number of undifferentiated and mature cells over time in culture with G-CSF. As shown in Fig. 2C, expression of the activated intracellular domain of mNotch1 allowed continued expansion of undifferentiated cells. After 5–6 days, the majority of cells in the control groups demonstrated some degree of granulocytic maturation and very few cells remained undifferentiated (Fig. 2C Middle); there was an average seven-fold decrease in the absolute number of undifferentiated cells (<15% of the original number of cells plated) during this time. In contrast, the lines expressing mNotch1-ICΔOP demonstrated a progressive expansion of undifferentiated cells (Fig. 2C Bottom); by day 6 the average number of undifferentiated cells in these cultures was nearly 2000% of the original number of cells plated.
Therefore, we find that in addition to inhibiting differentiation, expression of mNotch1-ICΔOP permits continued proliferation of cells in an undifferentiated state. However, expression of mNotch1-ICΔOP does not stimulate proliferation per se; proliferation of mNotch-ICΔOP clones stimulated with G-CSF was less than that of cells maintained in IL-3 alone (compare with Fig. 2C Top and Bottom). The average proliferation of clones expressing the various retroviral constructs was comparable to 32D WT cells when maintained in IL-3 (not shown). Thus, expression of mNotch-ICΔOP does not enhance proliferation, but merely permits it to continue under conditions that normally result in its cessation. In addition, the proliferation of undifferentiated cells in G-CSF was not due to development of IL-3 independence, since the mNotch1-ICΔOP clones continued to require IL-3 for survival past a few days (data not shown).
Expression of the Activated Intracellular Domain of mNotch2 has No Effect on Granulocytic Differentiation of 32D Cells.
To further confirm the specificity of the effects of mNotch1-ICΔOP expression on the differentiation of 32D cells, we compared G-CSF-induced granulocytic differentiation of 32D cells expressing mNotch1-ICΔOP to cells expressing the corresponding region of mNotch-2 (mNotch2-ICΔOP). The use of mNotch2-ICΔOP permits a comparison between the effects of two very similar proteins, both containing the myc epitope tag. Sequence analysis of the mNotch2 construct predicted a protein product 98.7% identical to that of rat Notch2 (25) and comparable levels of mNotch1- and 2-ICΔOP protein expression were verified by Western blot (Fig. 3A). In addition, functional activity of the mNotch2-ICΔOP construct was verified in two other assays (R.K., unpublished observations). As shown in Fig. 3B, 32D clones expressing mNotch2-ICΔOP showed granulocytic differentiation comparable to that seen with the parental 32D cells and control clones (see Fig. 2A), in contrast to the cells expressing mNotch1-ICΔOP, which, as in other experiments, remained predominantly undifferentiated. These results provide additional confirmation that the effects seen in cells expressing mNotch1-ICΔOP are specific for Notch1 rather than a general effect of the myc tag or cdc10/ankyrin repeats.
Figure 3.
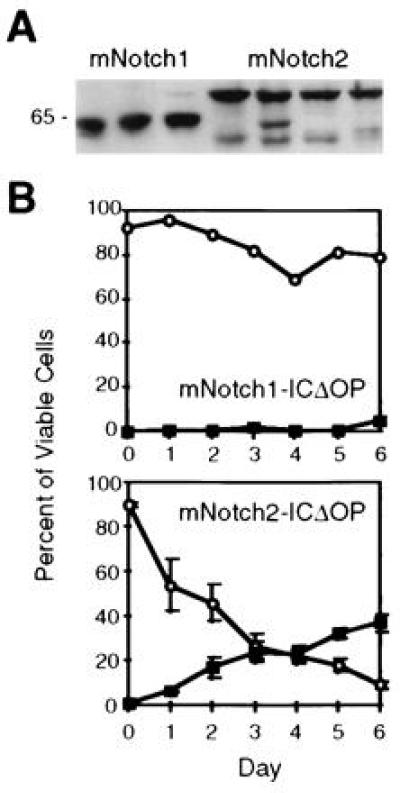
Comparison of mNotch1- and mNotch2-ICΔOP expression and effect on granulocytic differentiation of 32D cells. (A) Western blot of three representative mNotch1-ICΔOP and four mNotch2-ICΔOP 32D clones. Each lane contained a total cell lysate representing 5 × 106 cells electrophoresed through a 10% acrylamide gel, transferred to nitrocellulose, and immunostained using the 9e10 (α-myc) monoclonal antibody. The clones shown include those used for the experiment depicted in B. (B) G-CSF-induced granulocytic differentiation. In this experiment, three individual clones expressing mNotch2-ICΔOP, a representative mNotch1-ICΔOP clone, four LXSN-MT control clones, and the parental 32D WT cells were evaluated for differentiation in response to 50 ng/ml G-CSF and plotted as the relative percentages of viable cells remaining undifferentiated (○) or showing morphologic characteristics of mature granulocytes (▪). Differentiation of the mNotch2 clones was comparable to the 32D WT and LXSN-MT controls (not shown, but comparable to Fig. 2A).
DISCUSSION
We have demonstrated that expression of an activated form of mNotch1 inhibits G-CSF-induced granulocytic differentiation of 32D myeloid progenitor cells. 32D clones expressing the active region of the intracellular domain of mNotch1 continue to proliferate primarily as undifferentiated cells, in contrast to parental 32D cells and clones expressing control constructs, which terminally differentiate into mature granulocytes when stimulated with G-CSF. These findings are consistent with the general model emerging from studies of Notch function in other vertebrate systems (13–15), where expression of similar activated forms of Notch has been shown to inhibit cell type-specific differentiation.
Members of the Notch family are thought to mediate cell fate decisions by inhibiting or delaying differentiation along a particular pathway, leaving cells competent to respond to subsequent inductive signals and thus capable of adopting an alternate fate. Signaling through the Notch pathway is thought to occur through cell–cell interactions in which Notch functions as a receptor; ligand binding to the extracellular domain appears to modulate signal transduction through the intracellular domain, resulting in inhibition of differentiation (for reviews, see refs. 3–5). The intracellular signal transduction pathway of Notch involves nuclear factors and there is some evidence that the intracellular domain of Notch is proteolytically cleaved and translocated to the nucleus (28). Direct interactions of the intracellular domain of Notch with nuclear proteins such as Drosophila Suppressor of Hairless [Su(H)] (29), the mammalian homolog of Su(H), RBP-Jκ/CBF (30, 31), and NFκB (32) have been documented. The result of the interaction of Notch and Su(H) or CBF appears to be regulation of lineage-specific genes, most likely indirectly through increased expression of negative regulators such as Drosophila Enhancer of Split [E(spl)] and the mammalian homolog, Hairy Enhancer of Split (HES) (33–35).
Based on the molecular mechanisms implicated in signaling through the Notch pathway in other systems, we hypothesize that Notch functions at each maturation branch point during hematopoietic differentiation to inhibit expression of lineage-specific genes. The specific genes regulated by Notch in hematopoietic cells are unknown, but are likely to represent those required for the next step in the differentiation of a given cell. Given that 32D cells represent a heterogeneous population of myeloid progenitors at various stages of maturation, the genes ultimately regulated may vary from cell to cell; thus, the phenotype of individual clones would be expected to reflect the maturational stage of the cell when Notch became activated. While it is unlikely that Notch activation results in a discrete block in differentiation, it is likely that Notch functions through a common signal transduction pathway involving the regulation of transcription factors [such as Su(H) and E(spl)-like molecules]. Considering the frequent association of Notch with the regulation of basic helix-loop-helix proteins (1, 15, 35–38), it is potentially of interest that the basic helix-loop-helix protein Id also inhibits granulocytic differentiation of 32D cells (39). Studies are in progress to identify molecules interacting with Notch and genes regulated by Notch activity in 32D cells; these studies should help elucidate the signaling pathway of Notch in myeloid differentiation.
In the studies presented here, we have demonstrated that an activated form of mNotch1 inhibits granulocytic differentiation of myeloid progenitors. It remains possible that ligand activation of the native Notch1 molecule will not result in this same inhibitory effect in normal hematopoietic progenitors in vivo. However, when considered together with the inhibitory effects of Notch signaling through extracellular ligand binding in other systems, our results suggest that Notch functions in a similar way in hematopoietic cells.
In the context of the normal hematopoietic microenvironment, the effects of Notch activation are likely to be complex due to the influence of multiple cell–cell signaling pathways, the effects of various cytokines, and the capacity of individual hematopoietic cells to express lineage-specific genes. In addition, stimulation of Notch activity in vivo is likely to be transient, allowing cells to receive subsequent signals and adopt alternate differentiative fates. In the present studies, 32D cells constitutively expressing activated Notch would not be expected to demonstrate an alternate cell fate because of the lack of alternative signals and because unregulated expression of activated Notch is likely to result in continuous inhibition of differentiation. In this case, the “alternate” cell fate may be self-renewal of relatively undifferentiated progenitors. We propose that in the context of the normal hematopoietic microenvironment, regulation of Notch activity through extracellular ligand binding will permit only those cells maintaining the highest relative levels of Notch expression to remain multipotent.
Our findings in 32D cells are similar to the effects of unregulated expression of hNotch1 (TAN-1) in T lymphocytes. The predicted protein product resulting from the translocation involving the hNotch1 (TAN-1) gene in some T-lymphoblastic leukemias lacks most of the extracellular domain of hNotch1, suggesting that unregulated activation of the intracellular domain of hNotch1 may contribute to the development of these malignancies. Recent studies demonstrating the development of T-cell leukemias and lymphomas in mice transplanted with bone marrow expressing similar truncated Notch1 molecules support this notion (40). The effects of unregulated Notch activation in leukemic cells may be to inhibit further differentiation, permitting uncontrolled expansion of immature cells at the expense of normally maturing cells, leading to the malignant phenotype. These effects are comparable to those we observed with constitutive expression of the activated intracellular domain of mNotch1 in 32D cells; in this case, mNotch1-expressing 32D cells continue to proliferate in an undifferentiated state rather than undergoing granulocytic differentiation in response to G-CSF.
The process of hematopoiesis has many similarities to other developmental processes in which pluripotent stem cells give rise to progeny that undergo proliferation and maturation to result in mature cells of different lineages. Members of the Notch family are known to be general mediators of cell fate determination in many of these developmental processes and have highly conserved structural and functional features both in invertebrate and vertebrate systems. We have speculated that members of the Notch family are also involved in mediating cell fate decisions during hematopoiesis. The findings presented here, demonstrating that expression of an activated form of mNotch1 inhibits granulocytic differentiation of 32D myeloid progenitors, support the view that Notch is capable of influencing cell fate determination in hematopoietic cells.
Hematopoiesis is a complex process that is highly regulated through the interactions of various signaling pathways involving positive and negative, soluble and cell-bound, factors (for reviews, see refs. 41–44). The intersection of Notch signaling with these other pathways is also likely to be very complex and highly regulated. In addition, four mammalian Notch genes (9–12, 25, 36, 45–49) [and several putative Notch ligands (5, 50, 51)] have now been identified and at least three of these are expressed in hematopoietic cells (L.A.M., unpublished work). It is possible that the different Notch molecules have distinct functions in hematopoietic cells; our observation that an active form of mNotch1, but not the corresponding form of mNotch2, inhibits G-CSF-induced granulocytic differentiation of 32D cells supports this hypothesis. Keeping these issues in mind, we speculate that in the appropriate context, multiple Notch family members function during hematopoiesis to inhibit differentiation, leaving some cells multipotent at each maturational stage; depending on the subsequent signals encountered and the relative levels of Notch expressed, these cells may then choose to adopt a specific cell fate, or to again self-renew, maintaining a multipotent phenotype. Such a role for Notch in hematopoiesis has broad implications for the regulation of the numbers and types of cells produced during hematopoietic differentiation. Further studies to define the role of Notch molecules in hematopoiesis may provide new insights into disorders of hematopoiesis, including cytopenias and hematopoietic malignancies, and may contribute to achieving the potential for stem cell expansion.
Acknowledgments
This manuscript is dedicated to the memory of Hal Weintraub, colleague, mentor, and friend. We would like to thank G. Rovera for the 32D C13 cell line, D. Miller for the pLXSN vector, and E. C. B. Milner for helpful discussions. L.A.M. and R.K. are Fellows and D.I.K.M. is a Scholar of the Leukemia Society of America. A.B. is the recipient of a Postdoctoral Fellowship from Comisio Intradepartamental de Recerca i Innovacio Tecnologica, Generalitat de Catalunya, Spain. This work was supported in part by funds from The James McDonnell Foundation, National Institutes of Health Grant P30 HD28834 through the University of Washington Child Health Research Center to L.A.M., and National Institutes of Health Grant 5RO1HC 48790 to D.I.K.M.
Footnotes
References
- 1.Artavanis-Tsakonas S, Simpson P. Trends Genet. 1991;7:403–408. doi: 10.1016/0168-9525(91)90264-q. [DOI] [PubMed] [Google Scholar]
- 2.Greenwald I, Rubin G M. Cell. 1992;68:271–281. doi: 10.1016/0092-8674(92)90470-w. [DOI] [PubMed] [Google Scholar]
- 3.Fortini M E, Artavanis-Tsakonas S. Cell. 1993;75:1245–1247. doi: 10.1016/0092-8674(93)90611-s. [DOI] [PubMed] [Google Scholar]
- 4.Muskavitch M A T. Dev Biol. 1994;166:415–430. doi: 10.1006/dbio.1994.1326. [DOI] [PubMed] [Google Scholar]
- 5.Artavanis-Tsakonas S, Matsuno K, Fortini M E. Science. 1995;268:225–232. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- 6.Wharton K A, Johansen K M, Xu T, Artavanis-Tsakonas S. Cell. 1985;43:567. doi: 10.1016/0092-8674(85)90229-6. [DOI] [PubMed] [Google Scholar]
- 7.Yochem J, Greenwald I. Cell. 1989;58:553–563. doi: 10.1016/0092-8674(89)90436-4. [DOI] [PubMed] [Google Scholar]
- 8.Coffman C, Harris W, Kintner C. Science. 1990;249:1438–1441. doi: 10.1126/science.2402639. [DOI] [PubMed] [Google Scholar]
- 9.Weinmaster G, Roberts V J, Lemke G. Development (Cambridge, UK) 1991;113:199–205. doi: 10.1242/dev.113.1.199. [DOI] [PubMed] [Google Scholar]
- 10.Franco del Amo F, Smith D E, Swiatek P J, Gendron-Maguire M, Greenspan R J, McMahon A P, Gridley T. Development (Cambridge, UK) 1992;115:737–744. doi: 10.1242/dev.115.3.737. [DOI] [PubMed] [Google Scholar]
- 11.Kopan R, Weintraub H. J Cell Biol. 1993;121:631–641. doi: 10.1083/jcb.121.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellisen L W, Bird J, West D C, Soreng A L, Reynolds T C, Smith S D, Sklar J. Cell. 1991;66:649–661. doi: 10.1016/0092-8674(91)90111-b. [DOI] [PubMed] [Google Scholar]
- 13.Coffman C R, Skoglund P, Harris W A, Kintner C R. Cell. 1993;73:659–671. doi: 10.1016/0092-8674(93)90247-n. [DOI] [PubMed] [Google Scholar]
- 14.Nye J S, Kopan R, Axel R. Development (Cambridge, UK) 1994;120:2421–2430. doi: 10.1242/dev.120.9.2421. [DOI] [PubMed] [Google Scholar]
- 15.Kopan R, Nye J S, Weintraub H. Development (Cambridge, UK) 1994;120:2385–2396. doi: 10.1242/dev.120.9.2385. [DOI] [PubMed] [Google Scholar]
- 16.Struhl G, Fitzgerald K, Greenwald I. Cell. 1993;74:331–345. doi: 10.1016/0092-8674(93)90424-o. [DOI] [PubMed] [Google Scholar]
- 17.Fortini M E, Rebay I, Caron L A, Artavanis-Tsakonas S. Nature (London) 1993;365:555–557. doi: 10.1038/365555a0. [DOI] [PubMed] [Google Scholar]
- 18.Rebay I, Fehon R G, Artavanis-Tsakonas S. Cell. 1993;74:319–329. doi: 10.1016/0092-8674(93)90423-n. [DOI] [PubMed] [Google Scholar]
- 19.Roehl H, Kimble J. Nature (London) 1993;364:632–635. doi: 10.1038/364632a0. [DOI] [PubMed] [Google Scholar]
- 20.Lieber T, Kidd S, Alcamo E, Corbin V, Young M W. Genes Dev. 1993;7:1949–1965. doi: 10.1101/gad.7.10.1949. [DOI] [PubMed] [Google Scholar]
- 21.Robbins J, Blondel B J, Gallahan D, Callahan R. J Virol. 1992;66:2594–2599. doi: 10.1128/jvi.66.4.2594-2599.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jhappan C, Gallahan D, Stahle C, Chu E, Smith G H, Merlino G, Callahan R. Genes Dev. 1992;6:345–355. doi: 10.1101/gad.6.3.345. [DOI] [PubMed] [Google Scholar]
- 23.Milner L A, Kopan R, Martin D I, Bernstein I D. Blood. 1994;83:2057–2062. [PubMed] [Google Scholar]
- 24.Evan G I, Lewis G K, Ramsay G, Bishop M. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinmaster G, Roberts V J, Lemke G. Development (Cambridge, UK) 1992;116:931–941. doi: 10.1242/dev.116.4.931. [DOI] [PubMed] [Google Scholar]
- 26.Miller A D, Rosman G J. BioTechniques. 1989;7:980–990. [PMC free article] [PubMed] [Google Scholar]
- 27.Valtieri M, Tweardy D J, Caracciolo D, Johnson K, Mavilio F, Altmann S, Santoli D, Rovera G. J Immunol. 1987;138:3829–3835. [PubMed] [Google Scholar]
- 28.Kopan R, Schroeter E H, Nye J, Weintraub H. Proc Natl Acad Sci USA. 1996;93:1683–1688. doi: 10.1073/pnas.93.4.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fortini M E, Artavanis-Tsakonas S. Cell. 1994;79:273–282. doi: 10.1016/0092-8674(94)90196-1. [DOI] [PubMed] [Google Scholar]
- 30.Tamura K, Taniguchi Y, Minoguchi S, Sakai T, Tun T, Furakawa T, Honjo T. Curr Biol. 1995;5:1416–1423. doi: 10.1016/s0960-9822(95)00279-x. [DOI] [PubMed] [Google Scholar]
- 31.Hsieh J J, Henkel T, Salmon P, Robey E, Peterson M G, Hayward S D. Mol Cell Biol. 1996;16:952–959. doi: 10.1128/mcb.16.3.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guan E, Wang J, Laborda J, Norcross M, Baeuerle P A, Hoffman T. J Exp Med. 1996;183:2025–2032. doi: 10.1084/jem.183.5.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lecourtois M, Schweisguth F. Genes Dev. 1995;9:2598–2608. doi: 10.1101/gad.9.21.2598. [DOI] [PubMed] [Google Scholar]
- 34.Bailey A M, Posakony J W. Genes Dev. 1995;9:2609–2622. doi: 10.1101/gad.9.21.2609. [DOI] [PubMed] [Google Scholar]
- 35.Jarriault S, Brou C, Logeat F, Schroeter E H, Kopan R, Israel A. Nature (London) 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- 36.Stifani S, Blaumueller C M, Redhead N J, Hill R E, Artavanis-Tsakonas S. Nat Genet. 1992;2:119–127. doi: 10.1038/ng1092-119. [DOI] [PubMed] [Google Scholar]
- 37.Tata, F. & Hartley, D. A. (1993) Development (Cambridge, U.K.) Suppl., 139–148. [PubMed]
- 38.Jennings B, Decelis J, Delidakis C, Preiss A, Bray S. Development (Cambridge, UK) 1995;121:3745–3752. [Google Scholar]
- 39.Kreider B L, Benezra R, Rovera G, Kadesch T. Science. 1992;255:1700–1702. doi: 10.1126/science.1372755. [DOI] [PubMed] [Google Scholar]
- 40.Pear W S, Aster J C, Scott M L, Hasserjian R P, Soffer B, Sklar J, Baltimore D. J Exp Med. 1996;183:2283–2291. doi: 10.1084/jem.183.5.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Metcalf D. Blood. 1993;82:3515–3523. [PubMed] [Google Scholar]
- 42.Ogawa M. Blood. 1993;81:2844–2853. [PubMed] [Google Scholar]
- 43.Orkin S H. Curr Biol. 1995;7:870–877. doi: 10.1016/0955-0674(95)80072-7. [DOI] [PubMed] [Google Scholar]
- 44.Shivdasani R A, Orkin S H. Blood. 1996;87:4025–4039. [PubMed] [Google Scholar]
- 45.Lardelli M, Lendahl U. Exp Cell Res. 1993;204:364– 372. doi: 10.1006/excr.1993.1044. [DOI] [PubMed] [Google Scholar]
- 46.Lardelli M, Dahlstrand J, Lendahl U. Mech Dev. 1994;46:123–136. doi: 10.1016/0925-4773(94)90081-7. [DOI] [PubMed] [Google Scholar]
- 47.Larsson C, Lardelli M, White I, Lendahl U. Genomics. 1994;24:253–258. doi: 10.1006/geno.1994.1613. [DOI] [PubMed] [Google Scholar]
- 48.Sugaya K, Fukagawa T, Matsumoto K, Mita K, Takahashi E, Ando A, Inoko H, Ikemura T. Genomics. 1994;23:408–419. doi: 10.1006/geno.1994.1517. [DOI] [PubMed] [Google Scholar]
- 49.Uyttendaele H, Marazzi G, Wu G, Yan Q, Sassoon D, Kitajewski J. Development (Cambridge, UK) 1996;122:2251–2259. doi: 10.1242/dev.122.7.2251. [DOI] [PubMed] [Google Scholar]
- 50.Simpson P. Nature (London) 1995;375:736–737. doi: 10.1038/375736a0. [DOI] [PubMed] [Google Scholar]
- 51.Nye J S, Kopan R. Curr Biol. 1995;5:966–969. doi: 10.1016/s0960-9822(95)00189-8. [DOI] [PubMed] [Google Scholar]