

Abstract
The conserved organization of the Hox genes throughout the animal kingdom has become one of the major paradigms of evolutionary developmental biology. We have examined the organization of the Hox genes of the grasshopper, Schistocerca gregaria. We find that the grasshopper Hox cluster is over 700 kb long, and is not split into equivalents of the Antennapedia complex and the bithorax complex of Drosophila melanogaster. SgDax and probably also Sgzen, the grasshopper homologues of fushi-tarazu (ftz) and Zerknüllt (zen), respectively, are also in the cluster, showing that the non-homeotic Antp-class genes, “accessory genes,” are an ancient feature of insect Hox clusters.
Keywords: fluorescent in situ hybridization, pulsed-field gel electrophoresis, accessory genes, gene linkage
The Hox genes, first defined by a subset of homeotic mutations in flies, are now known to be present in diverse metazoan phyla. They are homeobox-containing genes, encoding transcription factors that specify the position of cells during early embryonic development. This role has been demonstated in chordates, arthropods, and nematodes, and is likely to have been acquired early in the evolution of the metazoa (1, 2).
Hox genes are clustered in the genome. A characteristic but puzzling feature of these clusters is that the order of the genes within the cluster generally parallels the position along the body where the genes are expressed. This “spatial colinearity” is most evident in chordates, where all the genes also lie in the same transcriptional orientation. Genes at the 3′ end of the clusters are expressed anteriorly, and genes at the 5′ end of the cluster are expressed posteriorly. This organization is replicated in each of the four Hox clusters present in the genomes of mammals, fishes, and birds. Although some Hox genes have been lost from the array present in individual clusters, no case has yet come to light where the overall organization of the cluster has been disrupted by rearrangement.
In vertebrates, colinearity extends also to the temporal sequence in which the genes are activated during embryogenesis. Anterior, 3′ genes are always activated before posterior, 5′ genes. Colinearity has led to the strong belief that the physical organization of the genes on the chromosome is significant for their normal role in development (3).
Outside of the chordates, the chromosomal organization of Hox genes has been characterized only in insects, and in one nematode, Caenorhabditis elegans. The nematode cluster contains only 4 genes (c.f., 13 in the presumed ancestral vertebrate cluster), and these 4 genes are present in 2 pairs, separated by an intervening sequence that contains many unrelated genes. The order of one gene pair has been inverted with respect to the expected colinear pattern (4). It seems likely that the C. elegans cluster displays a derived and secondarily simplified organization, particularly as the ancestral metazoan Hox cluster probably contained more than four Hox genes (5).
Among insects, the Drosophila Hox genes have been mapped most thoroughly. Their organization resembles that of the vertebrate clusters in that the genes show an overall pattern of spatial colinearity, but this is disrupted in several ways. Genes homologous to a single Hox cluster of chordates are split into two clusters, which are separated by about five chromosome divisions (approximately 7.5 Mb, or 5% of the genome). The Antennapedia complex (ANT-C) contains anterior genes that specify the identity of head and mouthpart segments, whereas the bithorax complex (BX-C) contains posterior genes that act in the thorax and abdomen. The ANT-C is further disrupted by the inverted transcriptional orientation of two homeobox genes, by the presence of homeobox genes that do not retain colinear expression, and by the presence within the cluster of genes wholly unrelated to the Hox genes (6).
Temporal colinearity is not apparent in Drosophila. Drosophila makes segments almost simultaneously along the whole length of its body axis. Anterior and posterior Hox genes are activated at much the same time, and there is no dependence of posterior genes on the prior activation of the anterior genes. Instead, each gene is activated by a unique combination of transcription factors that is generated at a particular position along the axis of the embryo, in a syncytial environment (7).
A further difference between vertebrate and Drosophila clusters lies in the organization of transcriptional and regulatory elements. The vertebrate clusters are compact (100–120 kb), and the individual transcription units are short (2). The Drosophila clusters are larger, with introns and intergenic regulatory regions that are exceptionally long compared with most other genes in this compact genome. The regulatory regions appear to be organized into discrete segment-specific domains that are largely autonomous (8).
These differences have prompted the suggestion that the vertebrate and Drosophila Hox clusters are now subject to very different structural and regulatory constraints, and more specifically, that colinearity is not, or is no longer, functionally significant in the Drosophila cluster. This view is supported by the observation that the Drosophila melanogaster bithorax cluster can be split experimentally without obvious deleterious effect (9, 10), and that different rearrangements that split the Hox cluster have been fixed in two Drosophilid lineages (11).
The organization of the Hox cluster has been examined in two other non-Drosophilid insects, the beetle, Tribolium castaneum, and the moth, Bombyx mori (12, 13). The Hox genes of both of these insects map within 2–3 centimorgans and, hence, are not split as far apart as the two Drosophila complexes. However, no molecular linkage data are yet available for the Hox genes of these insects.
We have chosen to examine the organization of the Hox cluster in the grasshopper Schistocerca gregaria, to determine whether the genes have remained clustered and whether the organization is compact, like that of vertebrates, or extensive, like Drosophila. Furthermore S. gregaria forms all segments posterior to the cephalic segments progressively, in a cellular environment (the “short germ” mode of development). Thus, it contrasts sharply with the simultaneous generation of segments in the syncytial blastoderm of Drosophila (the “long germ” mode of development) (14). If these different modes of development impinge on the regulation and organization of the Hox genes, then this may be most evident in a comparison of the two extreme germ types represented by Drosophila and grasshopper.
We have used chromosomal Fluorescent In Situ Hybridization and CHromosomal InterPhase Staining [FISH and CHIPS (15)], combined with Southern blot analysis and pulsed-field gel electrophoresis (PFGE), to examine the organization of the Hox genes in S. gregaria. We show that the grasshopper cluster is not split into the equivalents of the fly ANT-C and BX-C. The single S. gregaria cluster is at least 700 kb long, but probably no longer than 2 Mb. It is located on either chromosome 9 or 10. The SgDax gene, and probably the Sgzen gene, homologues of ftz and zen respectively, are in the grasshopper cluster.
MATERIALS AND METHODS
FISH and CHIPS.
Spreads of metaphase and interphase nuclei were made from grasshopper embryos at the 35% stage of development (16). The embryos were dissected from their eggs under locust embryo saline (150 mM NaCl/3 mM KCl/2 mM CaCl2/1 mM MgSO4/5 mM TES (N-tris[hydroxymethyl]methyl-2-aminoethanesulfonic acid/2-([2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino)ethanesulfonic acid) (Sigma)), treated with 0.02% colcemid (Sigma) for 1 hr, swollen in 1% sodium citrate for 15 min, and fixed in 3:1 ethanol/acetic acid for a minimum of 1 day at 4°C. The fixed embryos were mushed up on a siliconized coverslip in a drop of fresh 45% acetic acid, and squashed between the coverslip and a slide. The slide was submerged in liquid nitrogen until the bubbling stopped, and the coverslip flicked off. The spreads were dehydrated in 95% ethanol (3 times for 5 min each), and stored in a dry, air-tight, dark box.
Probes were made by nick translation according to standard procedures (17), to incorporate either biotin or digoxigenin. The genomic clones used are described in Dawson (18) (Scr and histone marker), Tear et al. (19) (abd-A), Kelsh et al. (20) (Abd-B), Dawes et al. (21), (SgDax), and Procunier and Smith (22) (ribosomal marker). The histone clone derives from Schistocerca americana, and the ribosomal RNA clone from D. melanogaster. All the others are from S. gregaria.
Slides were dehydrated through ethanol (5 min in each of 70%, 70%, 95%, 95%, 100% solutions) and then air-dried, followed by denaturation in 70% formamide (70°C, 2 min), and a second dehydration through an ethanol series (the first 70% ethanol being ice-cold). Denatured probe (100–150 ng), in 15 μl of hybridization mix (50% formamide/600 mM NaCl/10% dextran sulphate/2× saline sodium citrate (SSC)/1 mM EDTA/12 mM tris, pH 7.6/5× Denhardt’s solution/100 μg/ml sheared salmon sperm DNA), was sealed over the chromosome spreads with a 22 × 22 mm coverslip and rubber cement. Hybridization at 42°C was carried out overnight. The rubber cement was then removed and the coverslips washed off in 2× SSC at 42°C. The slides were washed in 50% formamide in 2× SSC (42°C, 2 times for 5 min each), followed by 2× SSC (42°C, 2 times for 5 min each), and then 4× TNFM (37°C, for 30 min and 5 min each) (4× SSC/0.05% Tween 20/5% non-fat milk powder). The sites of hybridization were detected with avidin–fluorescein isothiocyanate (Vector Laboratories) (1:500 dilution in 4× TNFM for 1 hr at 37°C), followed by 4× TNFM washes (42°C, 3 times for 5 min each), biotinylated-anti-avidin (room temperature, 20 min), 4× TNFM washes (42°C, 2 times for 5 min each), and a second layer of FITC–avidin (room temperature, 20 min). The slides were then washed in 4× TNFM (42°C, 2 times for 5 min each), followed by 4× SSC with 0.05% Tween 20 (room temperature, 2 times for 5 min each), and then 1× PBS (room temperature, 5 min), before counterstaining the chromatin with Hoechst solution (1 μg/ml in 1× PBS, at room temperature for 30 min). After a final rinse with 1× PBS (room temperature for 5 min) the spreads were mounted in Vectashield mountant (Vector Laboratories).
The signals were visualized on a Zeiss Axiophot microscope and photographed on Kodak GoldII ASA400 film. Brightness and contrast were adjusted in Adobe photoshop 3.
Genomic DNA Preparation.
Testicular follicles were dissected from anesthetized adult male grasshoppers (from a population maintained in the Zoology Department, Cambridge University) under locust embryo saline. The follicles were broken up in ice-cold 1× PBS in a Dounce homogenizer with a loose-fitting plunger, and the cells separated from the rest of the follicular debris by passing through two layers of 37-μm Nitex mesh. The cells were pelleted for 5 min at 2750 × g at 4°C, and resuspended in 1 ml of ice-cold 1× PBS. The washing was repeated a total of three times. The cells were finally resuspended in L buffer (17), and warmed to 42°C for 10 min, prior to mixing with an equal volume of 2% GIBCO Ultrapure LMP agarose in water, at 42°C. Aliquots (90 μl) were pipetted into Bio-Rad plug moulds and allowed to set at 4°C for over 30 min. Each 90-μl plug contained 40–200 μg of DNA (corresponding to testicular follicle cells from 6–8 adult males). The set plugs were washed 2 times for 24 hr with 50 vol of L buffer containing 1 mg/ml proteinase K and 1% lithium dodecyl sulfate, followed by 2 times for 1 hr with 50 vol of TE buffer (pH 7.6) and 40 μg/ml phenylmethylsulfonyl fluoride, and stored in TE buffer (pH 7.6) at 4°C.
DNA Digests.
One whole plug was used for a single digest per gel lane. Plugs were washed twice for 30 min in TE buffer (pH 7.6) at 4°C, and then equilibrated with 10 vol of 1× restriction enzyme buffer at 4°C for more than 1 hr. The buffer was replaced with 3 vol of 1× restriction enzyme buffer and 80 units of restriction enzyme, and allowed to equilibrate at 4°C for more than 2 hr prior to incubation at the appropriate digestion temperature. During the incubation the enzyme and buffer were replaced, and the incubation continued for a total of approximately 20 hr. The plugs were then equilibrated with 0.5× TBE running buffer on ice for more than 2 hr.
PFGE.
The Hoefer HULA PFGE system was used. The running conditions were 180 V/11°C/40 hr/20–120-sec ramped pulse time, with the current being turned off while the gel reoriented.
Southern Blot Analysis.
Southern blotting was carried out as described in ref. 17, with denaturing solution (1.5 M NaCl/0.5 M NaOH) as the transfer buffer, onto Hybond N+ membrane (Amersham).
The probes were made by PCR. For Abd-B the probe was 558 bp of intronic sequence; for abd-A the probe was 243 bp, including part of the homeobox and a short stretch downstream; for Scr the probe was 2 kb, including the homeobox; for SgDax the probe was 660 bp of the cDNA 5′ of the homeobox; for Sgzen the probe was 508 bp, including most of the homeobox and some sequence upstream (23); and for eve the probe was 370 bp, including most of the homeobox and some upstream sequence. Primer information is available on request. Approximately 10 ng of template DNA, from a previous cold PCR reaction, was used in a reaction with [32P]dCTP (50 μCi; 1 Ci = 37 GBq) (DuPont), and 200 μM each of dATP, dGTP, and dTTP. The amplification conditions were 95°C for 5 min, 72°C while the Taq polymerase is added, (15 cycles of 94°C for 30 sec/50°C for 30 sec/72°C for 2 min), and 72°C for 5 min. Unincorporated nucleotides were removed with a Stratagene NucTrap push column, following the manufacturer’s instructions.
Southern blot hybridization was carried out as described in ref. 17. Hybridization was for a minimum of 20 hr. Washes were at high stringency (0.1× SSPE/0.1% SDS at 65°C). Results were visualized on a PhosphorImager (Molecular Dynamics) after a 4–5 day exposure.
We were limited to using short probes because of the high frequency of repetitive sequences in the S. gregaria genome. This, together with the large size of the grasshopper genome (approximately 3 times that of human), necessitates overloading the gel to obtain a detectable signal. The resultant smiling of the bands on the pulsed field gel limits the accuracy of the size estimates to ±50 kb (estimated from several filters).
RESULTS
The S. gregaria Homologues of Scr, abd-A, Abd-B, and ftz Hybridize to the Same Short Chromosome. S.
gregaria has a haploid karyotype of 11 chromosomes, distinguishable by size into three distinct groups [numbers 1–3 long, 4–8 medium, and 9–11 short (24)]. The major nucleolar organizers are localized on chromosomes 3 and 6 (25), which correspond to the major sites of hybridization of a probe against ribosomal RNA genes (Fig. 1a). Tandem arrays of histone genes are on chromosome 8, according to estimates of chromosome lengths and agreement with previous assessments (P. Bella, personal communication).
Figure 1.
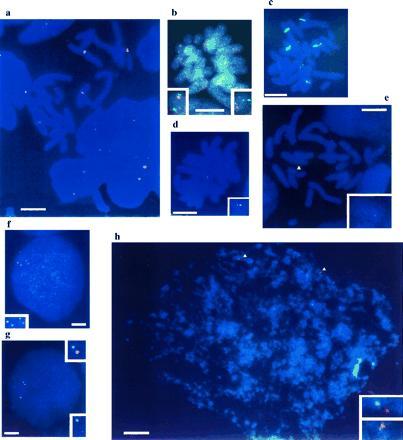
The tight linkage of grasshopper Scr, abd-A, Abd-B, and SgDax, as revealed on metaphase and interphase spreads (blue). (a) Metaphase chromosomes surrounded by interphase nuclei with the major ribosomal gene loci (red). (b) Metaphase with homologous chromosomes showing two Scr (green) signals and one Abd-B (red) signal each. The Abd-B signal in the right inset is faint because it is in a slightly different focal plane from the other signals. (c) Metaphase with Scr (red) and histone and ribosomal markers (green). (d) Metaphase with a single chromosome showing two abd-A (red) signals and one SgDax (green) signal. (e) Metaphase with a single chromosome (yellow arrowhead) bearing two abd-A (red) signals and one SgDax (green) signal. (f) Interphase with two Scr (red) and two SgDax (green) signals. (g) Interphase with two SgDax (green) and two abd-A (red) signals. (h) Interphase with two abd-A (red) and two Scr (green) signals (yellow arrowheads). Insets show enlargements of the signals. (Bars = 10 μm.)
A genomic fragment from the S. gregaria Sex combs reduced gene hybridizes in situ to a single site on one of the short chromosomes that does not carry either histone or rRNA markers (Fig. 1c). Length measurements show that this is not the shortest chromosome, but either chromosome 9 or 10.
SgDax, abd-A, and Abd-B map close to Scr on the same chromosome. We have shown this by double-labeling experiments with probes for two different single-copy genes, one labeled with biotin and the other with digoxigenin, to enable simultaneous detection (Fig. 1 and data not shown). In D. melanogaster, Scr and ftz lie in the ANT-C, whereas abd-A and Abd-B are in the BX-C. Thus, genes homologous to both ANT-C (Scr, SgDax) and BX-C (abd-A, Abd-B) are closely linked in S. gregaria.
To estimate the molecular distance between these sites, the same probes were hybridized to spread chromatin from interphase nuclei. For each pair tested (Scr/abd-A, Scr/SgDax, SgDax/abd-A, SgDax/Abd-B), the sites of hybridization were separated by no more than an average of 2.6 μm. On similar spreads of human cells, this would correspond to a molecular distance of less than 2 Mb (26–28). This rough estimate is in reasonable agreement with the data from pulse field gels presented below.
The Minimum Size of the Grasshopper Cluster Is 700 kb.
To estimate the size of this Hox cluster more accurately, we digested genomic DNA with rare cutting retriction enzymes, separated the fragments by PFGE, and used Southern blot hybridization to locate the fragments carrying Hox genes.
The restriction enzyme AscI produces fragments of 100–700 kb containing grasshopper Hox genes. Despite their size, none of these AscI fragments contained more than a single one of the genes for which we have probed (Fig. 2). This means that the minimum size of the grasshopper cluster is 700 kb (summing the fragments containing Scr, SgDax, and abd-A, but excluding the fragments containing the presumed 3′ and 5′ genes).
Figure 2.
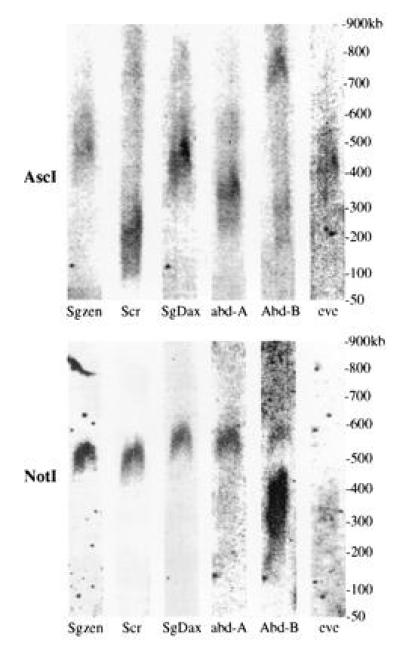
Grasshopper Hox gene Southern bands from PFGE. Different Hox gene probes produce bands of different sizes from each other with AscI (Sgzen = 375–425 kb; Scr = 100 kb; SgDax = 350–400 kb; abd-A = 225–300 kb; Abd-B = 650–700 kb). With NotI Sgzen and Scr probes give bands in the same place, at about 450 kb, whereas SgDax, abd-A, and Abd-B probes all give bands at 500–550 kb. The Abd-B probe produces two bands, at 350 kb and 500–550 kb, presumably due to some kind of polymorphism (see text). The eve probe produces AscI and NotI bands of different sizes from the Hox gene probes (AscI band = 400 kb, NotI band = 300 kb). The eve bands are very faint because S. gregaria DNA was probed with a Schistocerca americana sequence. The even-skipped (eve) NotI band is barely visible here, but is clear on other filters, which are not shown due to incomplete data for the other probes. A shorter exposure of the Abd-B/NotI experiment shows more clearly that the short Abd-B band is at 350 kb, and is longer than the eve band, at 300 kb. The AscI bands are all on the same filter, which was probed, stripped, and reprobed each time. Similarly, all of the NotI bands are on the same filter.
We also wanted to find rare-cutting restriction enzymes that produced bands containing more than one Hox gene. NotI appears to be such an enzyme. NotI produces a 500–550 kb band with SgDax, abd-A, and Abd-B probes, and a 450-kb band with the Scr probe and part of a cDNA-derived from the S. gregaria zen homologue, Sgzen (23) (Fig. 2). The Abd-B probe also hybridizes to a 350-kb NotI band. The origin of the additional Abd-B band is not clear, but is probably due to polymorphism. The same probe detected single strong bands of 4.5–20 kb in digests with each of eight different six-cutter enzymes, which suggests that the gene is not duplicated. The AscI data and the NotI data together are consistent with the possibility that SgDax, abd-A, and Abd-B are on the same 500–550-kb NotI fragment, with Sgzen and Scr on an adjacent 450-kb fragment (summarized in Fig. 3). These data do not rule out the possibility that the cluster is much larger than this.
Figure 3.

Summary of the pulsed field data showing the hypothesized linkage of Hox genes on the NotI fragments. None of the probes co-localize on the same AscI fragment. The linkage data for the homologous D. melanogaster genes is shown for comparison.
None of the above data prove that there is only a single grasshopper Hox cluster. The sequences used as probes may not be sufficiently conserved to hybridize at high stringency to paralogous Hox loci. However, two lines of evidence suggest that there is only a single Hox cluster. First, most short homeobox fragments from the Hox cluster genes hybridize to single bands on conventional Southern blots (data not shown). Second, degenerate primer PCR has provided no evidence for multiple distinct copies of single Hox classes, despite recovering three genes more than six times each (I. Millwood and K. Moyse, personal communication).
Grasshopper eve Is Not Closely Linked to Abd-B.
In vertebrates, both eve class homeobox genes are located adjacent to the 5′ end of the Hox clusters (corresponding to the site of the insect Abd-B gene). We used an eve cDNA obtained from S. americana (29) as a probe to see if eve is present on the same 650–700 kb AscI fragment as Abd-B; it is not (see Fig. 2). Because this AscI fragment does not contain abd-A, it probably extends well beyond the Hox cluster. However, the arrangement of the Abd-B end of the Hox cluster may be unusual (indicated by the two NotI bands), and so we cannot promote this conclusion firmly.
DISCUSSION
Clustered Hox genes are present in the genomes of many metazoa, and wherever it has been tested, these genes have been shown to specify the different behavior of cells at different positions along the body axis. However, despite this conservation, the structure of the Hox clusters differs considerably in each of the groups that have been examined. In vertebrates the genes are short and individual clusters are compact. Multiple, paralogous clusters are present in each genome (2). In Drosophila the genes are large, regulatory elements cover many kilobases, and the single cluster has been split by distinct inversion events in two different lineages (11). In one nematode the cluster is also split, several genes have probably been lost, and the two 3′ genes are inverted with respect to colinearity expectations (4, 5).
In part, these differences may reflect the changing functional organization of the Hox clusters. For example, it has been suggested that the vertebrate Hox genes are regulated by some mechanism that requires tight colinear clusters (30), and the vertebrate genes have shared and overlapping regulatory elements (31, 32). The Drosophila genes, however, seem largely autonomous, with little sharing of regulatory elements (8). It has also been suggested that Drosophila may exploit the time it takes to transcribe very long genes to coordinate developmental events over a timescale of hours (33, 34).
However, many differences between species must reflect frozen accidents of history that are of no functional consequence (e.g., some chromosome rearrangements) or of no direct relevance to the function of the Hox genes themselves (e.g., an increase of gene size by the accumulation and decay of transposable elements). There is no sure way to distinguish significant from trivial changes, but one way to assess proposed relationships between the structure and function of the Hox clusters is to determine whether the phylogenetic distribution of particular characteristics is consistent with their proposed function. Do independent rearrangements disrupt the Hox cluster in many arthropod lineages, but very rarely or never in chordates? Are long Hox genes unique characteristics of insects that develop very fast, or are they also found in insects that take days rather than hours to set up patterns of Hox gene regulation?
Our evidence suggests that the grasshopper Hox cluster is not split, at least between Scr and Abd-B. In this it resembles T. castaneum, and probably B. mori, the only non-Drosophilid insects to have been studied (12, 13). Thus, there is some suggestion that rearrangement within the cluster may be more easily tolerated in higher Diptera than in less derived insect species.
The grasshopper cluster is at least as large as the combined Drosophila ANT-C and BX-C (≈700 kb), and possibly as much as 2 Mb. We do not know the length of individual transcription units, but these cannot be as short as those in chordates. We have previously isolated phage clones that carry the 3′ (homeobox) exons of Scr, SgDax, abd-A, and Abd-B. In none of these can we recognize the 5′ exon of the gene, even though the Scr and SgDax clones extend more than 10 kb upstream from the homeobox exon. The large size of these genes, and of the whole cluster, suggests that S. gregaria contains extensive gene-specific regulatory elements, rather than the compact, overlapping elements of vertebrates.
Even if the genes are larger than those in Drosophila, it is unlikely that their rate of transcription is used to time developmental events. Embryonic development takes about 20 times longer in Schistocerca than in Drosophila (3 weeks at 29°C), so the time taken to transcribe even a 250-kb transcription unit (≈4 hr) would be but a small fraction of development.
In addition to the highly conserved homeotic genes of the Hox clusters, the grasshopper has homologues for at least two of the “accessory” homeobox genes in the Drosophila cluster, ftz and zen [SgDax and Sgzen, respectively (21, 23)]. Our mapping data show that the ftz homologue is certainly linked to the Hox cluster, and suggest that zen may be too. These genes are probably derived from ancestral Hox gene sequences, but are evolving more rapidly than the surrounding homeotic Hox genes, both with respect to sequence and expression. They appear to be doing so within the chromosomal context of the conserved and tightly regulated Hox genes.
Miller and Miles (35) suggested that eve may be in the ancestral Hox cluster, as it is linked to Hox genes in vertebrates and in a coral (but not in Drosophila). We have found no evidence to suggest that S. gregaria eve is tightly linked to the Hox cluster, even though its expression, in a uniform posterior domain, is rather “Hox-like” (29).
Acknowledgments
We thank Nigel Carter for the FISH and CHIPS protocol, Pepe Bella for assistance with Schistocerca cytology, Justin Ainscough for advice on PFGE, Nipam Patel for the S. americana eve cDNA clone, summer students Iona Millwood and Keith Moyse for allowing us to cite their PCR results, and Sandra Rylance for help with grasshopper butchery. This work was supported by the Wellcome Trust.
Footnotes
Abbreviations: ANT-C, Antennapedia complex; BX-C, bithorax complex; PFGE, pulsed-field gel electrophoresis.
References
- 1.Slack J M W, Holland P W H, Graham C F. Nature (London) 1993;361:490–492. doi: 10.1038/361490a0. [DOI] [PubMed] [Google Scholar]
- 2.McGinnis W, Krumlauf R. Cell. 1992;68:283–302. doi: 10.1016/0092-8674(92)90471-n. [DOI] [PubMed] [Google Scholar]
- 3.Duboule D, Morata G. Trends Genet. 1994;10:358–364. doi: 10.1016/0168-9525(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 4.Wang B B, Muller-Immergluck M M, Austin J, Tamar Robinson N, Chisholm A, Kenyon C. Cell. 1993;74:29–42. doi: 10.1016/0092-8674(93)90292-x. [DOI] [PubMed] [Google Scholar]
- 5.Balavoine G, Telford M J. Proc Natl Acad Sci USA. 1995;92:7227–7231. doi: 10.1073/pnas.92.16.7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindsley D L, Zimm G G. The Genome of Drosophila melanogaster. San Diego: Academic; 1992. [Google Scholar]
- 7.Akam M. Development (Cambridge, UK) 1987;101:1–22. [PubMed] [Google Scholar]
- 8.Averof M, Dawes R E D, Ferrier D E K. Semin Dev Biol. 1996;7:539–551. [Google Scholar]
- 9.Struhl G. Nature (London) 1984;308:454–457. [Google Scholar]
- 10.Tiong S Y K, Whittle J R S, Gribbin M C. Development (Cambridge, UK) 1987;101:135–142. doi: 10.1242/dev.101.1.135. [DOI] [PubMed] [Google Scholar]
- 11.Von Allmen G, Hogga I, Spierer A, Karch F, Bender W, Gyurkovics H, Lewis E. Nature (London) 1996;380:116. doi: 10.1038/380116a0. [DOI] [PubMed] [Google Scholar]
- 12.Beeman R. Nature (London) 1987;327:247–249. [Google Scholar]
- 13.Ueno K, Hui C-C, Fukuta M, Suzuki Y. Development (Cambridge, UK) 1992;114:555–563. doi: 10.1242/dev.114.3.555. [DOI] [PubMed] [Google Scholar]
- 14.Akam M, Dawes R E D. Curr Biol. 1992;2:395–398. doi: 10.1016/0960-9822(92)90313-y. [DOI] [PubMed] [Google Scholar]
- 15.Norby S. Nature (London) 1994;370:255. [Google Scholar]
- 16.Bentley D, Keshishian H, Shankland M, Toroian-Raymond A. J Embryol Exp Morphol. 1979;54:47–74. [PubMed] [Google Scholar]
- 17.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 18.Dawson I. Ph.D. thesis. Cambridge, U.K.: Cambridge Univ.; 1989. [Google Scholar]
- 19.Tear G, Akam M, Martinez-Arias A. Development Cambridge, UK) 1990;110:915–925. doi: 10.1242/dev.110.3.915. [DOI] [PubMed] [Google Scholar]
- 20.Kelsh R N, Dawson I A, Akam M. Development (Cambridge, UK) 1993;117:293–305. doi: 10.1242/dev.117.1.293. [DOI] [PubMed] [Google Scholar]
- 21.Dawes R E, Dawson I, Falciani F, Tear G, Akam M. Development (Cambridge, UK) 1994;120:1561–1572. doi: 10.1242/dev.120.6.1561. [DOI] [PubMed] [Google Scholar]
- 22.Procunier W S, Smith J J. Insect Mol Biol. 1993;2:163–174. doi: 10.1111/j.1365-2583.1993.tb00136.x. [DOI] [PubMed] [Google Scholar]
- 23.Falciani F, Akam M, Hausdorf B, Schröder R, Tautz D, Denell R, Brown S. Proc Natl Acad Sci USA. 1996;93:8479–8484. doi: 10.1073/pnas.93.16.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.John B, Hewitt G M. Chromosoma. 1966;20:155–172. [Google Scholar]
- 25.Fox D P, Carter K C, Hewitt G M. Heredity. 1973;31:272–276. doi: 10.1038/hdy.1973.83. [DOI] [PubMed] [Google Scholar]
- 26.Lawrence J B, Singer R H, McNeil J A. Science. 1990;249:928–932. doi: 10.1126/science.2203143. [DOI] [PubMed] [Google Scholar]
- 27.Trask B J. Trends Genet. 1991;7:149–154. doi: 10.1016/0168-9525(91)90378-4. [DOI] [PubMed] [Google Scholar]
- 28.van den Engh G, Sachs R, Trask B J. Science. 1992;257:1410–1412. doi: 10.1126/science.1388286. [DOI] [PubMed] [Google Scholar]
- 29.Patel N H, Ball E E, Goodman C S. Nature (London) 1992;357:339–342. doi: 10.1038/357339a0. [DOI] [PubMed] [Google Scholar]
- 30.Duboule, D. (1994) Development (Cambridge, U.K.) Suppl., 135–142.
- 31.Whiting J, Marshall H, Cook M, Krumlauf R, Rigby P W J, Stott D, Allemann R K. Genes Dev. 1991;5:2048–2059. doi: 10.1101/gad.5.11.2048. [DOI] [PubMed] [Google Scholar]
- 32.Boncinelli E, Simeone A, Acampora D, Mavilio F. Trends Genet. 1991;7:329–334. doi: 10.1016/0168-9525(91)90423-n. [DOI] [PubMed] [Google Scholar]
- 33.Shermoen A W, O’Farrell P H. Cell. 1991;67:303–310. doi: 10.1016/0092-8674(91)90182-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gubb D. Dev Genet. 1986;7:119–131. [Google Scholar]
- 35.Miller D J, Miles A. Nature (London) 1993;365:215–216. doi: 10.1038/365215b0. [DOI] [PubMed] [Google Scholar]