

Abstract
We recently reported that chlorophyll (Chl) strongly inhibits aflatoxin B1 preneoplasia biomarkers in rats when administered by co-gavage (Simonich et al., Carcinogenesis, 28:1294–1302, 2007). The present study extends this by examining the effects of dietary Chl on tumor development, using rainbow trout to explore ubiquity of mechanism. Duplicate groups of 140 trout were fed diet containing 224 ppm dibenzo[a,l]pyrene (DBP) alone, or with 1000 – 6000 ppm Chl, for 4 weeks. DBP induced high tumor incidences in liver (51%) and stomach (56%), whereas Chl co-fed at 2000, 4000 or 6000 ppm reduced incidences in stomach (to 29, 23 and 19%, resp., P<0.005) and liver (to 21, 28 and 26%, resp., P < 0.0005). Chlorophyllin (CHL) at 2000 ppm gave similar protection. Chl complexed with DBP in vitro (2Chl:DBP, Kd1 = 4.44 ± 0.46 μM, Kd2 = 3.30 ± 0.18 μM), as did CHL (Kd1 = 1.38 ± 0.32 μM, Kd2 = 1.17 ± 0.05 μM), possibly explaining their ability to inhibit DBP uptake into the liver by 61–63% (P<0.001). This is the first demonstration that dietary Chl can reduce tumorigenesis in any whole animal model, and that it may do so by a simple, species-independent mechanism.
Keywords: chlorophyll; dibenzo[a,l]pyrene; tumor incidence; bioavailability; liver cancer; stomach cancer
Introduction
Numerous chemicals purified from fruits and vegetables protect against carcinogenesis in experimental animal models (Wattenberg, 1990; Kohlmeier et al., 1995; Hayatsu et al., 1988; Dragsted et al., 1993). Often, these phytochemicals occur in edible plants at such low levels that doses sufficient for chemoprotection in animal models are not practically attained in a balanced human diet (Breinholt et al., 1995b). Because of its abundance in a green vegetable-rich diet, chlorophyll and its derivatives have attracted considerable attention as potential anti-carcinogens.
The anti-carcinogenic properties of chlorophyllin (CHL), a structural analogue of Chl, have been extensively reported. CHL is a food-grade, water-soluble derivative of chlorophyll that exhibits strong anti-mutagenic activity against a variety of carcinogens in prokaryotic and eukaryotic mutagenesis assays in vitro (Wu et al., 1994; Ong et al., 1986; Whong et al., 1988; Warner et al., 1991; Romert et al., 1992; Negishi et al., 1994). CHL cancer chemoprevention in vivo was first demonstrated in a rainbow trout study, where dietary CHL was shown to reduce aflatoxin B1 (AFB1)-induced DNA damage and hepatic tumor incidence with increasing CHL dose (Dashwood et al., 1991; Breinholt et al., 1995a). CHL subsequently proved to be similarly effective at blocking DNA adduct formation and tumor initiation in a variety of rodent tumor models as well as a trout multi-organ model (Dingley et al., 2003; Hasegawa et al., 1995; Guo et al., 1995b; Guo et al., 1995a; Park and Surh, 1996; Chung et al., 1999; Kim et al., 2000). Several possible mechanisms of CHL blocking have been proposed (reviewed in (Dashwood et al., 1998), including tight complex formation with the carcinogen and subsequent reduction of carcinogen bioavailability, inhibition of bioactivating enzymes, induction of detoxifying enzymes, in situ electrophile scavenging of the proximate carcinogen, and direct antioxidant activity. Most recently CHL was used in a randomized, double-blind, placebo controlled chemoprevention trial in rural China on human subjects unavoidably and chronically exposed to aflatoxin in their diet (Egner et al., 2001). Ingestion of 100 mg CHL at each meal for 3 months reduced the mean urinary level of aflatoxin-N7-guanine adducts by 55% compared to subjects taking placebo. Thus, initial discoveries in the lower vertebrate trout model (Dashwood et al., 1991; Breinholt et al., 1995a) were directly translatable to humans, and suggest that diet supplementation with CHL might substantially reduce human liver cancer risk from AFB1 exposure.
Chlorophyll (Chl), the parent compound of CHL, is readily available by consumption of green vegetables. Spinach leaves, for example, may be up to 2% (20,000 ppm) chlorophyll by dry weight. Chlorophyll is also a known anti-mutagen (reviewed in (Negishi et al., 1997)), and a weak inducer of mammalian phase 2 proteins in vitro that protect against oxidative damage (Fahey et al., 2005). A few whole animal studies have provided evidence that natural Chl might have cancer preventive properties in vivo. Harttig and Bailey (Harttig and Bailey, 1998) found that trout exposed via the diet for 2 weeks to 200 ppm dibenzo(a,l)pyrene (DBP) and 3000 ppm of several different Chl preparations had 66% mean inhibition of adduct formation relative to treatments with DBP alone. A similar concentration of dietary CHL produced nearly identical inhibition. In the rat colon, dietary spinach or an equimolar amount of Chl inhibited cytotoxicity and colonocyte proliferation induced by heme, a red meat component hypothesized to contribute to colon cancer risk (de Vogel et al., 2005; Sesink et al., 1999). Rats were fed diet supplemented with heme and a 2.4 fold molar excess of Chl, or spinach equaling that amount of Chl. Both the spinach and Chl supplementation abolished the nearly 8 fold and 2 fold respective increases in cytoxicity and colonocyte proliferation seen with the heme diet alone. In addition, the Chl-containing diet largely blocked formation of a cytotoxic heme metabolite (de Vogel et al., 2005). The authors speculated that green vegetables may decrease colon cancer risk from dietary heme through the protective effects of Chl.
Despite this promise, there appears to have been no whole-animal tumor study investigating the effects of dietary Chl on tumor response. The present study used the rainbow trout carcinogenesis model to compare the effects of dietary Chl and CHL against DBP multi-organ tumor development. The trout model was chosen in part because of its 40 year history of development as an effective, low-cost model in the investigation of cancer and its modulation by dietary factors (Dashwood et al., 1991; Breinholt et al., 1995a; Reddy et al., 1999; Dashwood et al., 1998; Harttig and Bailey, 1998; Sinnhuber et al., 1978; Hendricks et al., 1984; Lee, 1991; Bailey et al., 1996; Williams et al., 2003), and in part because we were specifically interested here to know if Chl protection might occur in lower as well as higher vertebrate models, by mechanisms largely species-independent and thus readily extrapolated to humans. Chl and CHL interactions with DBP in vitro and their effects on DBP bio-distribution in vivo were examined to explore mechanisms. These experiments addressed the possibility that chlorophylls may protect in part by reducing systemic uptake of the carcinogen, and that this might occur by molecular complex formation during co-exposure. We recently reported similar Chl protection against aflatoxin B1 DNA adduction and pre-neoplastic lesions when given by gavage in the rat (Simonich et al., 2007), supporting the idea of cross-species Chl protective mechanisms.
Materials and methods
Chemicals
Chlorophyllin (CHL), triethylene glycol (TEG) and tricaprylin were from Sigma Chemical Co. (St. Louis, MO). Dibenzo[a,l]pyrene (DBP) and 14C-DBP (55.9 mCi/mmole) was from the NCI Chemical Carcinogen Reference Standard Repository at Midwest Research Institute (Kansas City, MO). The purity and concentration of DBP was confirmed by absorbance in ethanol at 316 nm (ε316 = 4.75 x 104 M-1). The chlorin content of CHL was based on the manufacturer’s assay of 4.5% copper and assertion that all copper was present as copper-chlorins. Chlorophyll (Chl) was prepared as described below.
Preparation of chlorophyll
A comprehensive description of the Chl purification has been presented elsewhere (Jubert and Bailey, 2007). Briefly, organic spinach was purchased from a local supplier, washed with cold water, and freeze-dried after stem removal. The dried leaves were washed twice with petroleum ether (boiling point 30–60°C) and then extracted twice using methanol/petroleum ether (3:1, v/v). The combined extracts were transferred to a separatory funnel and washed with saturated sodium chloride. The aqueous layer was extracted again with petroleum ether and the petroleum ether layers were combined and washed with saturated sodium chloride. The final extract was filtered and evaporated in vacuo (T < 30°C). The extracted product contained other pigments such as carotenoids as well as oils, fats and waxes derived from the spinach. This crude Chl extract (90% pure by HPLC) was further purified by counter current chromatography (CCC) using an Ito multilayer-coil separator-extractor (P.C., Potomac, MD). Analyses of CCC fractions were performed by HPLC and by proton nuclear magnetic resonance (1H-NMR). A minor impurity was noticeable at δ 5.40 consisting of 0.35 protons upon integration, and the only other extra peaks were the NMR solvent (δ 2.05) and the two signals due to H2O and deuterated water (δ 2.78 and 2.75). Absence of any residual solvents from the extraction/purification was verified by spectral analysis. Purity was estimated to be >95% compared to Chl-a standards (Sigma Chemical Co.) which were shown to be 90–92% pure based on spectroscopic measurements. The yield from 30 grams of freeze-dried spinach was 300 milligrams of Chl-a and 100 mg of Chl-b. Chl preparations used in all experiments were a recombined 3:1 mixture of Chl-a : Chl-b.
Preparation of test solutions
Concentrated stocks ( >1 mg/ml) of DBP were first prepared in dichloromethane and diluted to working concentrations in ethanol. Solutions of CHL were prepared in water. Chl is virtually insoluble in water, thus all solutions were prepared in ethanol.
Animals
Shasta strain rainbow trout were reared in the Sinnhuber Aquatic Research Laboratory of Oregon State University as published elsewhere (Sinnhuber et al., 1978). Trout were maintained on a semi-synthetic Oregon Test Diet (OTD) formula (Lee, 1991). This formulation has proven suitable for inclusion of test chemicals varying widely in terms of solubility (hydrophilic, hydrophobic, insoluble) or origin (plant-based or not, natural or synthetic). This approach avoids the potential limitation of poor plant digestibility by trout, which are not herbivores in nature. Trout are Pavlovian in feeding response, and rapidly consume all diet introduced into the tanks within seconds. This, coupled with the poor solubility of the solid OTD formulation and high water exchange rates in our system, assures negligible opportunity for test agents to escape the diet or to interact with each other within the tank water.
Tumor studies in trout
For tumor studies, DBP and Chl were dissolved separately in the fish oil component of OTD. (Note: DBP is a potent carcinogen; it was handled, stored and disposed of in compliance with NIH and Oregon State University guidelines for extreme hazard class carcinogens.) CHL was dissolved in the water component of the OTD formulation. Since commercially available CHL is a mixture of chlorins and inorganic salts (Dashwood, 1997), the dietary CHL concentration was corrected to the actual copper chlorophyllin content (51.3%) of the lot used. According to the supplier, the remaining constituents are inorganic salts, primarily NaCl that have a negligible effect on tumor response (Reddy et al., 1999). This CHL formulation was chosen to mimic those in earlier in vitro, animal, and human intervention studies (Hayatsu et al., 1988; Breinholt et al., 1995b; Egner et al., 2001). It should be noted that natural chlorophylls with magnesium, CHL with copper, and simple protoporphyrins lacking any metal ion, were equally effective in protecting against DBP-DNA adduction in vivo (Harttig and Bailey, 1998). Chl, CHL and DBP are light-sensitive compounds, therefore, all diets were prepared under subdued lighting. Diets were prepared every two weeks and stored in the dark at −20 °C until one day prior to feeding when they were moved to 4°C.
Trout were fed control OTD from swim-up to 20 weeks of age. Duplicate tanks of 140 trout fry averaging 2.0 grams each were exposed via the diet for 4 weeks. The DBP, CHL and Chl concentrations were calculated on a dry diet weight basis, and diet was fed at the rate of 10% body weight per week. The study included 4 control groups: OTD alone (group 1), OTD + 2000 ppm Chl (group 2) or 2000 ppm CHL (group 3) and OTD containing 224 ppm DBP (group 4). Additional groups received diets containing 224 ppm DBP plus Chl at 1000 ppm (group 5), 2000 ppm (group 6), 4000 ppm (group 7) or 6000 ppm (group 8). Group 9 received DBP plus 2000 ppm CHL as a positive control for chemoprevention in this trout model (Reddy et al., 1999). Group 10 was fed 224 ppm DBP for the four week initiation phase, then fed 2000 ppm CHL (post-initiation) for the remainder of the 9 month study. Quantities of highly purified Chl were too limited to permit a similar prolonged post-initiation study. After 2 weeks and 4 weeks of exposure diet, 15 fish per tank were removed and killed by MS-222 overdose to obtain livers and stomachs for possible future biomarker assessments. (We note that dietary treatment with CHL (Reddy et al., 1999; Harttig and Bailey, 1998) or Chl (Harttig and Bailey, 1998) is already known to strongly suppress target organ DBP-DNA adducts in trout and that biomarker determination was not repeated for this single DBP-dose study, rather a more rigorous 12,000-animal dose-dose matrix study is under way to quantify the interrelationships between DBP dose, Chl dose, target organ DBP-DNA adduction, and eventual tumor outcome (Bailey et al., personal communication)). The remaining 110 fish per tank were fed OTD for 36 weeks, after which time all fish were killed and liver and stomach tumor development were quantified. As previously reported for 200 ppm DBP (Reddy et al., 1999), this protocol induced no DBP-related toxicity (see Table 1).
Table I.
Modulation of DBP tumorigenesis by dietary Chl in rainbow trouta
| Treatment group | Initial fish no. | Final fish no. | Weight (g)b | Tumor incidence (%) | Liver tumor typesd (%) | Tumor multiplicitye | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Liver | Stomach | SBc | HCC | HCA | MC | CCA | Liver | Stomach | SB | ||||
| Control diet (OTD) | 280 | 218 | 90 ± 27 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2000 ppm Chl | 280 | 210 | 91 ± 25 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2000 ppm CHL | 280 | 214 | 89 ± 25 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 224 ppm DBP | 280 | 212 | 90 ± 26 | 51 | 56 | 10 | 64 | 31 | 5 | 0 | 2.60 | 1.62 | 1.17 |
| 224 ppm DBP + 1000 ppm Chl | 280 | 211 | 95 ± 29 | 34** | 58 | 16 | 60 | 27 | 11 | 2 | 1.79* | 1.79 | 1.14 |
| 224 ppm DBP + 2000 ppm Chl | 280 | 211 | 91 ± 25 | 21* | 29* | 4 | 67 | 26 | 7 | 0 | 1.87* | 1.40* | 1.00 |
| 224 ppm DBP + 4000 ppm Chl | 280 | 211 | 97 ± 32 | 28* | 23* | 9 | 63 | 22 | 15 | 0 | 2.06* | 1.30* | 1.82 |
| 224 ppm DBP + 6000 ppm Chl | 280 | 217 | 96 ± 25 | 26* | 19* | 7 | 50 | 40 | 9 | 1 | 2.06 | 1.33* | 1.00 |
| 224 ppm DBP + 2000 ppm CHL | 280 | 212 | 88 ± 26 | 21* | 26* | 8 | 55 | 33 | 12 | 0 | 2.01* | 1.24* | 1.04 |
| 224 ppm DBP, then 2000 ppm CHLf | 280 | 208 | 91 ± 25 | 54 | 64 | 38* | 62 | 30 | 7 | 1 | 2.35 | 1.82 | 1.20 |
Thirty-six week tumor responses. All treatments had duplicate tanks for which the data were combined. Thirty fish from each dose were removed on day 14 and day 28, for future biomarker examinations. Groups significantly different from the DBP-alone group are indicated with an asterisk (*P < 0.0005; **P < 0.005). Due to space constraints, variances are not shown. An equal number of immature male and female trout were included in the study, and there were no gender differences in tumor outcome.
Mean weight of duplicate tanks ± variance
SB, swim bladder.
Relative percentage of each tumor type compared with the total tumors observed. HCC, hepatocellular carcinoma; HCA, hepatocellular adenoma; MC, mixed hepatocellular/cholangiocellular carcinoma; CCA, cholangiocellular adenoma.
Mean tumors per tumor-bearing fish (SD values omitted to conserve space). Groups significantly different from the DBP-alone group are indicated with an asterisk (*P < 0.05). The data for liver represent apparent multiplicity (see Methods).
Post-initiation 2000 ppm dietary CHL, begun after the DBP exposure period and continued for the duration of the study.
Tumor histology
Tissues were examined under a dissecting scope for gross tumors (≥0.5mm diameter), fixed in Bouin’s solution and processed by routine histological procedures. Numerous tumor studies in trout over the past 40 years have shown that 100% of stomach and swimbladder tumors, and >95% of liver tumors are surface-oriented outgrowths that are easily detected at gross necropsy. One slide from each organ having one or more suspect tumors at necropsy was prepared for histology. Tumors were classified according to criteria established previously by Hendricks et al. (Hendricks et al., 1984) for liver neoplasms and Hendricks et al. (Hendricks et al., 1995) and Bailey et al. (Bailey et al., 1996) for stomach and swimbladder neoplasms. Percentages of the different histological types of liver neoplasms were calculated from the total number of each type divided by the total number of all hepatic neoplasms in the group. Apparent tumor multiplicity was calculated by dividing the total number of tumors observed grossly by the number of tumor-bearing fish. This endpoint is termed apparent tumor multiplicity because not every lesion in organs exhibiting multiple lesions at gross necropsy was examined histologically in this 2800 animal study.
Molecular complex formation in vitro
The potential for DBP to form a non-covalent complex with CHL or highly purified Chl was assessed by quenching of DBP fluorescence described previously in detail (Reddy et al., 1999). Dissociation constant (Kd) determinations were carried out in non-buffered triethyleneglycol (pH 8.0) which served as the DBP-Chl co-solvent for the in vivo studies of bioavailability, and provided appropriate linearity of DBP fluorescence and sensitivity for monitoring quenching. The initial concentration of DBP (substrate) was 1.18 μM in a 3 ml stirred quartz cuvette. CHL or Chl was added in 0.4 μM increments up to 5.2 μM CHL or 10 μM Chl, with negligible increase in assay volume from ligand additions. Fluorescence was monitored at 425 ± 8 nm with excitation at 318 ± 8 nm, and was recorded 2 minutes after each ligand addition on a SLM 8000 photon counting spectrofluorometer. The fluorescence quenching data were iteratively fitted to 2:1 CHL: DBP and 2:1 Chl : DBP models of binding stoichiometry, as previously described (Reddy et al., 1999).
Redox activity of Cu-CHL in vitro by low-temperature electron spin resonance (ESR)
Spectral measurements of CHL (0.5 mM) were carried out in 0.1 M phosphate buffer (pH 7.4), with or without 2 mM ascorbate and/or 10 mM bathophenanthroline disulphonate (BCS), a specific Cu(I) chelating agent. ESR spectra were recorded using a Varian E109 Century Series spectrometer at low temperature (77 K). The sample was placed in a standard quartz ESR tube prior to freezing. The sides of the tube were then warmed slightly to allow the frozen sample to slide into a finger Dewar filled with liquid nitrogen for acquisition of spectra.
Effect of Chl or CHL on DBP biodistribution kinetics
One hundred and eighty trout of 15–40 g body weight were fasted for 5 days before test sample gavage to minimize reflex regurgitation of test samples (trout held at 12oC take several days to digest a meal and thus are not starved during fasting as rodents would be). Solutions of 14C-DBP (200 μM DBP, 50 μCi/umole) alone, with 2 mM CHL (as Cu-chlorins), or with 2 mM Chl were prepared in triethylene glycol (TEG), a suitable co-solvent for compounds of wide-ranging solubilities such as CHL and Chl, with no observed toxicity to trout in vehicle gavages. The 10:1 molar ratio of CHL or Chl to DBP used in the gavage solutions was based on the dissociation contants for CHL-DBP and Chl-DBP complexation in TEG determined as described above, and calculated to assure >99% complexation of the DBP by CHL. Trout were lightly anesthetized one at a time in a weak solution of MS-222 until no longer swimming but still vigorously ventilating. Individual fish were weighed and gavaged at 1 μl/g body weight with the appropriate test solution using a 5 cm blunt-tipped feeding needle on a 50 μl Gastight Hamilton microsyringe (Reno, NV). Gavaged trout were held head-up for 10 s and placed in 3L of 12oC water for 10 minutes. At the end of 10 minutes, a 2 ml sample of the water was collected for scintillation counting as an estimate of short-term reflex regurgitation from each fish following gavage, and the trout were moved to a large tank for the duration of the experiment. Trout (N = 10, each data point) were killed by MS-222 overdose and the liver, stomach and blood collected at 1, 3, 6, 12, 24 and 72 hrs after gavage. Blood (approx. 0.4 ml) was collected from the caudal vein using a 21 gauge heparinized Vacutainer (Becton Dickinson, Franklin Lakes, NJ) and stored at –20oC until analysis. Liver and stomach were collected into 20 ml glass scintillation vials and frozen at –20oC until analysis. Blood samples (200 μl) were decolorized by the dropwise addition of 1 ml of fresh hydrogen peroxide, incubation overnight at room temperature, and subsequent incubation at 60oC for 1h to drive off oxygen. The liver and stomach samples were briefly homogenized in a mixture of 2 ml Soluene-350 (Perkin Elmer, Boston, MA) and 1 ml ethanol, and incubated at 60oC for several hrs to completely solubilize the tissue. Liver samples were decolorized as above. Stomach samples were decolorized with 4 ml hydrogen peroxide overnight, and purged of oxygen by incubation at 60oC for 2 hours. All of the blood and liver samples were mixed with 15 ml Hionic Fluor (Perkin Elmer) liquid scintillation fluid, and radioactivity was counted in a Beckman 7500 Liquid Scintillation counter. One ml of solubilized stomach was mixed with 10 mls Hionic Fluor and counted. The initial gavage dose each fish received was calculated by subtracting the short-term regurgitation measure recorded for each fish at 10 minutes post-gavage from the administered dose. Tissue responses from N = 10 fish per time point were averaged and the standard error of the mean reported.
Statistical methods
Statistics were performed on the logit of liver and stomach tumor incidence. Liver and stomach tumor incidences from duplicate tanks in each experimental diet were assumed to be binomially distributed. However, the variance in liver tumor incidences of the DBP + 6000 ppm Chl tanks was somewhat overdispersed (P = 0.115). Therefore, a quasi-likelihood χ2analysis allowing overdispersion relative to the binomial model was used to compare liver tumor incidences among the diets. The variance in stomach tumor incidences was not overdispersed (P = 0.291) and comparison among the diets was made using χ2 analysis assuming the data were binomially distributed. Analyses were performed using SAS version 9.1 (SAS Institute, Inc.). Differences in DBP bioavailability over time were compared from graphs of ‘% of dose detected’ vs. ‘hrs. after gavage’, using area under the curve (trapezoidal rule) determined with SAS version 9.1. Comparison of AUC between treatments over time required fitting a model that accommodated heterogeneity of the variance within a treatment over time. The model was fit using the Mixed procedure in SAS version 9.1 with the Kenward-Roger adjustment to account for estimation of multiple variance components (Kenward and Roger, 1997).
Results
Chl and CHL effects on DBP-induced tumorigenicity
Treatment with 224 ppm dietary DBP for 4 weeks induced a strong multi-organ tumor response 9 months later (Table 1). Average tumor incidences in liver and stomach were 15% and 8% higher, respectively, than observed in our previous study with a 200 ppm DBP treatment (Reddy et al., 1999). The primary target organ, under the conditions tested, was the stomach, followed by liver and swimbladder. A 10% tumor incidence in the swimbladder was lower than the 30% incidence observed from our previous study (Reddy et al., 1999), which diminished sensitivity to detect tumor inhibition in this organ. Stomach and swimbladder tumors were all benign papillary adenomas, as observed previously with DBP (Reddy et al., 1999). Liver tumor phenotypes were also as observed previously for DBP, with hepatocellular carcinomas and adenomas averaging >90% of liver tumors (Reddy et al., 1999).
Co-exposure to dietary Chl at the lowest dose (1000 ppm) reduced liver tumor incidence significantly (Table 1), whereas co-exposure to higher Chl doses (2000, 4000, 6000 ppm) significantly reduced tumor incidence in stomach as well as liver. In this study there was no apparent trend toward increasing protection with 2000–6000 ppm Chl. Co-exposure to 2000 ppm dietary CHL served as a positive control group for chemoprotection, and strongly reduced liver and stomach tumor incidence as previously observed (Reddy et al., 1999). By contrast, 2000 ppm dietary CHL begun after the DBP exposure period (post-initiation) and continued for the 9 month duration of the study, did not suppress tumor incidence in any organ. Instead it produced significant (P < 0.001) promotion of swimbladder tumors, and an apparent but non-significant (P = 0.066) elevation of stomach tumors. Tumor multiplicity (apparent multiplicity in liver) was only moderately affected by CHL or Chl co-exposure. In the liver, weak reduction of apparent multiplicity was significant (P < 0.05) for 1000 – 4000 ppm Chl, but not for 6000 ppm Chl. In the stomach the 2000 – 6000 ppm Chl co-exposures gave weak but significant (P < 0.05) reductions in multiplicity. The 2000 ppm CHL co-exposure produced similar reductions in liver and stomach tumor multiplicity, whereas post-initiation CHL did not influence multiplicity.
Molecular complex formation in vitro
Reddy et al (1999) reported that CHL complexed non-covalently in a 2:1 ratio of CHL:DBP binding with an overall dissociation constant (Kd1,2) of 1.59 μM in tetrahydrofuran. However, in vivo experiments necessitated the use of a more biologically compatible co-solvent in which CHL, Chl and DBP were soluble at concentrations ≥2 mM. TEG proved to be an effective amphipathic solvent with no observed toxicity to trout after gavage administration. To determine if CHL and Chl formed a non-covalent complex with DBP in TEG, and of sufficient stability to limit DBP uptake or metabolic activation, we monitored quenching of DBP fluorescence in solution upon incremental additions of CHL or Chl. Titration of DBP in TEG with CHL or Chl in 0.4 μM increments resulted in quenching of the DBP fluorescence spectrum between 400 and 500 nm as previously observed for CHL (Reddy et al., 1999) with the exception that the first addition of CHL slightly but reproducibly enhanced DBP fluorescence (data not shown), and the first addition of Chl did not change DBP fluorescence (Figure 1A).
Fig. 1.
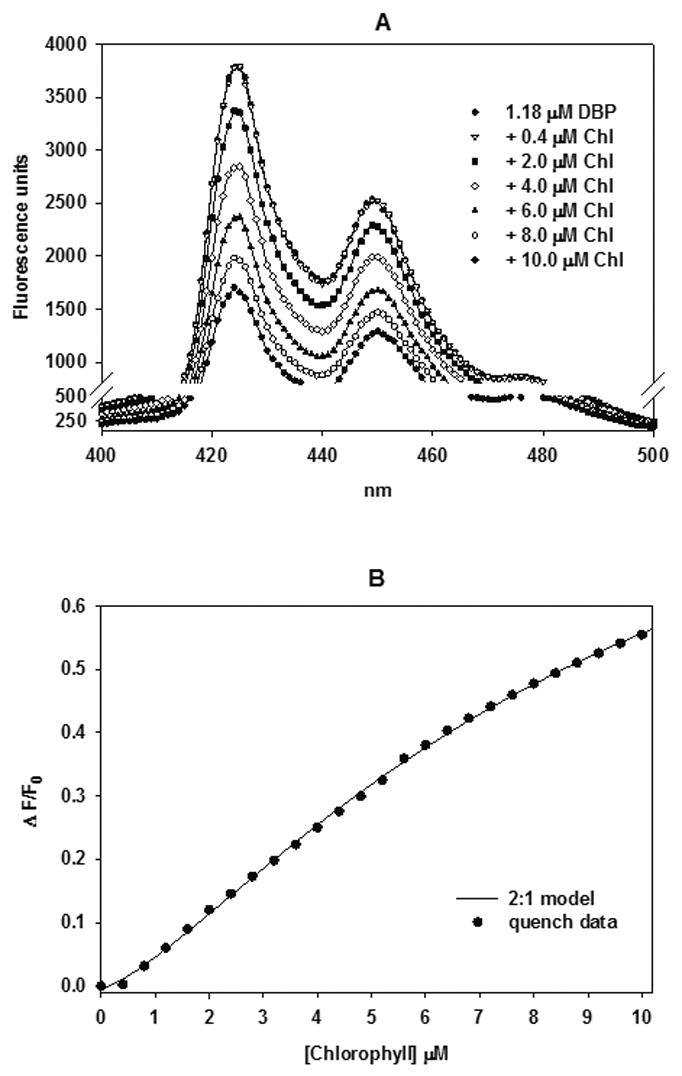
Spectrofluorometric titration of DBP with chlorophyll (Chl). (A) Effect of Chl on the DBP emission spectrum from 400 to 500 (± 8) nm (excitation 318 ± 8 nm) with DBP (substrate) concentration at 1.18μ. Chl (ligand) was added in 0.4 μM increments up to 10 μM (some titrations omitted from the figure for clarity) and the spectrum was recorded 2 minutes after each addition. (B) Quantification of Chl quenching of DBP fluorescence at 425 ± 8 nm recorded from the above spectra. Data were normalized by converting fluorescence units to ΔF/F0 and the data was fitted to the 2Chl:DBP complexation model.
To quantify CHL and Chl quenching of DBP fluorescence, the fractional fluorescence change (ΔF/F0) at 425 nm emission with each titration was plotted (Figure 1B CHL data not shown) and a model for a 2:1 ratio of CHL or Chl to DBP binding was constructed as described (Reddy et al., 1999). The 2:1 model allowed for quenching differences between the intermediate (1:1) and final (2:1) complexes. The total fluorescence of the binding assay was calculated as:
Where p1 = 1.09 and p2 = 0.56 are fluorescence enhancements, estimated from the binding response, for the complexes CHL-DBP and CHL2-DBP, respectively, and F is the molar fluorescence of DBP. Fitting of the data set to the 2:1 model (not shown) yielded Kd1 = 1.38 ± 0.32 μM, Kd2 = 1.17 ± 0.05 μM, (r2 = 0.9994). The residuals (data not shown), showed a weakly significant (P = 0.03) deviation from randomness corresponding to CHL concentrations between 1.2 and 2.8 μM. The goodness of fit, and hence the derived fitted values for Kd1 and Kd2 depended on the estimated values of p1 and p2. The lowest estimates of p1 = 1.07 and p2 = 0.48, for example, yielded much less precise estimates of Kd1 = 0.85 ± 4.51 μM, Kd2 = 1.96 ± 6.12 μM (r2 = 0.9968). We accept the derived best fitted values for Kd1 = 1.38 ± 0.32 μM, Kd2 = 1.17 ± 0.05 as reasonable estimates of CHL-DBP complex stability in TEG for the purposes of our in vivo studies.
Chl interaction with DBP in vitro was examined by a similar protocol. Fitting of the data set to the 2:1 model is shown in Figure 1B and yielded Kd1 = 4.44 ± 0.46 μM, Kd2 = 3.30 ± 0.18 μM, (r2 = 0.9996). The residuals also showed a weakly significant (P = 0.04) trend toward non-randomness corresponding to some of the lower Chl concentrations. The goodness of fit, and hence the derived fitted values for Kd1 and Kd2 were optimized when p1 = 0.88 and p2 = 0.17. We accept the derived best fitted values for Kd1 = 4.44 ± 0.46 μM, Kd2 = 3.30 ± 0.18 μM as reasonable estimates of Chl-DBP complex stability in TEG.
Chl and CHL effects on DBP biodistribution
In the trout model, available evidence suggests that CHL complexation with the mycotoxin aflatoxin B1 and consequent reduction of AFB1 bioavailability to the liver constitutes a primary route of chemoprotection (Breinholt et al., 1995b; Breinholt et al., 1999; Hayashi et al., 1999). The subsequent study demonstrating CHL-mediated reduction in urinary AFB1-DNA repair adducts in humans unavoidably exposed to AFB1 in their diets (Egner et al., 2001) is consistent with such a species-independent mechanism. The purpose of the present pharmacokinetic study was to determine if CHL and/or Chl might also modulate uptake and bio-distribution of DBP, a potent carcinogen in the polyaromatic hydrocarbon class. In this study, the ratio of CHL and Chl to DBP was chosen to assure >99% DBP complexation in the gavage mix. As seen in Figure 2 (B and C), CHL was quite effective at reducing the amount of DBP that appeared in the blood and liver, respectively following co-gavage. By area under the curve comparisons, CHL reduced blood DBP burden by 54% (P < 0.01) and liver DBP burden by 61% (P < 0.001) within the 72 hr observation period. Chl co-gavage was equally effective, and reduced blood DBP levels by 47% (P < 0.01) and hepatic DBP levels by 63% (P < 0.001), in accord with its ability to complex strongly with DBP in vitro.
Fig. 2.
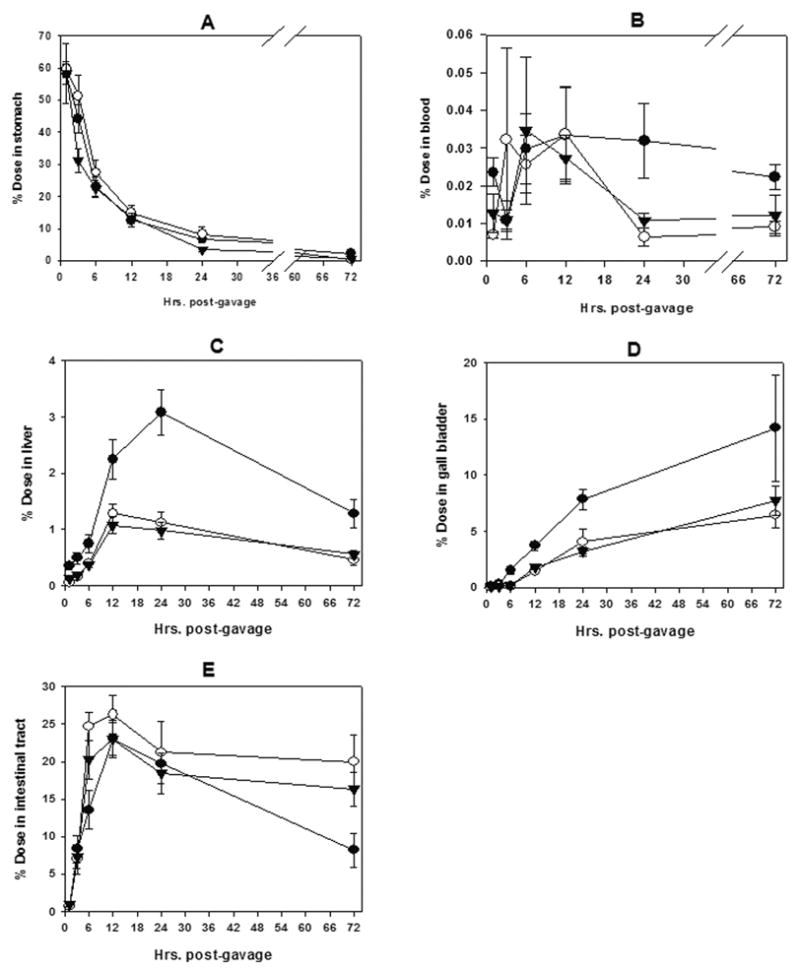
Pharmacokinetics of 200 μM [14C]-DBP following oral gavage treatment of 0.02 μCi/g body weight. Chemoprotective treatments included 2 mM chlorophyllin or 2 mM chlorophyll. Ten fish were killed at each time point after gavage Samples of each tissue were individually collected, processed and evaluated by liquid scintillation counting for 14C activity. Data from the pyloric cecae and the lower intestine were combined into one compartment termed the intestinal tract. ● = DBP, ○ = DBP + chlorophyllin, ▾ = DBP + chlorophyll. Each data point represents mean ± SE of the 10 samples.
Post-hepatic bile accumulation in the gall bladder (Figure 2D) of the fasted trout also indicated a similar cumulative reduction of DBP equivalents by Chl (52%) and CHL (53%) to that organ by the 72 hr time point (P < 0.01). CHL or Chl co-gavage did not appear to have a major effect on the retention times of DBP equivalents in the stomach (Figure 2A), but did significantly restrict uptake of DBP from the intestinal tract (Figure 2E). In the intestinal tract, area under the curve comparisons indicated that CHL and Chl co-gavage resulted in 27% (P < 0.01) and 15% (P < 0.05), respectively greater retention of DBP equivalents than the DBP-only treatment.
We note that the maximum DBP content in the blood and liver, seen between 12 and 24 hours post-gavage, represented only 0.033% and 3.1%, respectively of the initial dose. This indicates that overall bioavailability of oral DBP in the trout model was low compared with aflatoxin (Hayashi et al., 1999). We also note that at the 1hr time point an average of 42% of the gavage dose was absent from the stomach for each treatment. Since the regurgitation measured at 10 minutes was less than this (range 5%–35% of the dose among all fish), it is apparent that some additional regurgitation occurred over the next 50 minutes. Individual fish data could not be corrected for the un-quantified losses to regurgitation and limited metabolism between 10 minutes and 1 hour post-gavage. This became an additional source of inter-individual variation contributing to the error bars in Figure 2, which were large but not sufficiently so to obscure the substantial and significant reduction of DBP uptake and bioavailability by Chl or CHL co-treatment.
Redox activity of Cu-CHL in vitro
The potential for carcinogen inactivation by the Cu(II) component of Cu-CHL is often suggested as a potential detoxication mechanism. However, current evidence indicates that the Cu(II) is tightly bound under gastric simulation conditions (Ferruzzi et al., 2002), and it is unable to photoinduce dye reduction in vitro under conditions where the Zn derivative does so (Hidaka, 1985). We assessed the redox activity of the Cu(II) component in chlorophyllin using a low temperature ESR method. As seen in Figure 3, CHL has a characteristic ESR spectrum at low temperature (77K), showing the typical pattern of a square-planar Cu(II) complex. No decrease of the ESR signal intensity was observed after addition of ascorbate (2 mM), indicating that the Cu(II) in chlorophyllin can not be reduced to Cu(I) state (which has no ESR signal) by ascorbate. Even when bathophenanthroline disulphonate (BCS) was present together with ascorbate, the Cu (II) in chlorophyllin still could not be reduced to Cu(I). It should be noted that the shape of the ESR signal in the presence of BCS is different, which might be due to de-segregation of chlorophyllin by BCS. These ESR results support previous indications that Cu(II)-CHL is unable to express appreciable redox activity in vitro, and is thus unlikely to detoxify procarcinogens by such a mechanism in vivo.
Fig. 3.

Low-temperature ESR spectra of chlorophyllin with or without ascorbate and/or bathophenanthroline disulphonate (BCS). ESR measurements were carried out in 0.1 M phosphate buffer, pH 7.4, at 77K as described in Materials and Methods. The reaction mixtures contained 0.5 mM chlorophyllin, 2 mM ascorbate, or 2 mM ascorbate plus 10 mM BCS.
Discussion
Chl chemoprevention of DBP tumorigenesis in the trout model
An earlier pilot experiment suggested some ability of Chl to protect against liver tumors when delivered artificially, by co-injection with AFB1 into trout embryos (Dashwood et al., 1998). That suggestion is now confirmed and extended by the present study, which is the first, to our knowledge, to demonstrate inhibition of tumor development by dietary Chl in any whole animal model.
This finding is reinforced by our recent study showing similar protection by Chl as well as CHL against AFB1 DNA adduction and pre-neoplastic lesions in the rat when given by gavage (Simonich et al., 2007). The rat study clearly indicated that complex-mediated reduction of aflatoxin uptake in the rat was likely the dominant mechanism of chlorophyll chemoprevention. Our rationale for that claim was the dramatic reduction in urinary output of aflatoxin-DNA adducts in the rats that received chlorophyll plus aflatoxin, and the concomitant, dramatic increase of aflatoxin equivalents in the feces. The administration of chlorophyll with the aflatoxin effectively restricted aflatoxin to the rat GI tract. This simple, well established pharmacokinetic relationship was mirrored in the present trout model where, despite low DBP bioavailability, chlorophyll clearly restricted most of the DBP dose to the GI tract and prevented or retarded its appearance in the blood and liver. The combined data from the trout and rat models indicate that dietary or oral treatment with natural chlorophyll can provide potent chemoprotection against two important classes of carcinogen, through mechanism(s) operational in lower as well as higher vertebrates.
The reduction in tumor incidence afforded by 2000 ppm Chl against DBP-induced tumorigenesis was essentially equal to the protection from the same dose of CHL in both the liver and stomach. Tumor multiplicities for the liver and stomach reflected the same trend as incidence reductions, though not to the same magnitude. Since multiplicities in the DBP-only controls were low, reductions in multiplicity were only weakly significant (P < 0.05) and thus, not very useful end-points for assessing multi-organ Chl efficacy in this study. Interestingly, the present study provided no evidence for significant additional reduction of tumor incidence in either organ with 4000 or 6000 ppm Chl doses, which may be due to use of a non-optimal carcinogen dose (Pratt et al., 2006). A 12,000-animal Chl-DBP dose-dose matrix study is now in progress to examine biomarker and tumor dose-response issues in greater detail.
When CHL was limited to post-initiation dietary exposure, the observed stomach and swim bladder tumor incidences increased by 8% (P = 0.066) and 28% (P <0.001), respectively over the positive control incidence. This finding supports previous evidence that CHL chemoprevention is not without some potential risk. In particular, protocol-dependent post-initiation promotion by CHL has previously been reported in the rat, where 0.1% CHL in the drinking water after initiation increased the incidence of 1,2-dimethylhydrazine (DMH)-induced colon tumors from 10 to 47% (Nelson, 1992). Significantly increased colon tumor multiplicity was also observed in the rat following post-DMH initiation treatment with 0.001% CHL in the drinking water (Xu et al., 2001). However, 0.01% or 0.1% CHL in the same experiment had no effect on multiplicity, nor did any of the three CHL doses affect the multiplicity of 2-amino-3-methylimidazo[4,5-f]quinoline- (IQ; a heterocyclic amine carcinogen) induced colon tumors. In the one study where post-initiation effects of Chl on colon carcinogenesis were examined, Chl suppressed rather than promoted azoxymethane- (a metabolite of DMH) and IQ-induced aberrant crypt foci (Blum et al., 2003). This limited evidence suggests that natural Chl may provide protective potency equivalent to CHL without off-setting risk, and thus may be superior to CHL as a choice for chemoprevention in humans.
Mechanisms of Chl chemoprevention
Current evidence suggests that one mode of CHL chemoprevention of aflatoxin carcinogenesis in vivo is via complex formation with the carcinogen resulting in reduced bioavailability to target organs {Breinholt, 1995 #60;Breinholt, 1999 #45;Hayashi, 1999 #87;Simonich, 2007 #283}. Prior to the present study, however, there was no evidence to indicate that DBP uptake in vivo might also be modulated by CHL, or that Chl could directly modulate the bioavailability of any carcinogen. The present results indicate that Chl and CHL were each able to reduce carcinogen bioavailability to the liver when co-administered with DBP, with similar potency. The experiment was somewhat impaired by the innately low bioavailability of DBP in trout, which provided sub-optimal sensitivity for detection of CHL or Chl effects on rate of DBP uptake from the GI tract. Despite this, the liver, blood and gall bladder data clearly support the hypothesis that Chl, like CHL, affords chemoprotection at least in part through reduction of carcinogen bioavailability. The in vitro complexation studies are also consistent with the hypothesis that Chl- and CHL-mediated reduction in DBP bioavailability reflects formation of strong 2:1 complexes with DBP. However, the technical challenges of demonstrating Chl:DBP or CHL:DBP complexes within the stomach in the presence of food, make unambiguous proof of complex importance very difficult.
One alternative mechanism to explain systemic reduction in carcinogen bioavailability would be non-complex-mediated masking of carcinogen uptake, perhaps via transporter interaction. Additional protective mechanisms leading to altered carcinogen metabolism and less DNA damage within the target organ have also been suggested. These include degradation of the carcinogen or its proximate electrophile, cytochrome P450 enzyme inhibition, and phase II enzyme induction, all of which have been demonstrated to occur in vitro for CHL and/or Chl (Dingley et al., 2003; Fahey et al., 2005; Tachino et al., 1994; Sato et al., 1984; Yun et al., 1995). However, the relative importance of such mechanisms in vivo remains to be established (Dashwood et al., 1998), and in the case of enzyme induction, appears negligible (Dashwood et al., 1998; Breinholt et al., 1999; Simonich et al., 2007). Finally, the possibility that CHL-mediated redox destruction of procarcinogen in stored or freshly made diets may account for its apparent protective effects is not supported by our previous (Pratt et al., 2006) or present (Fig. 3) studies.
Chl - DBP complex formation in vitro
The in vitro complexation experiments described previously (Reddy et al., 1999) and herein for CHL-DBP indicate strong CHL-DBP complexation with an overall Kd < 2 μM, depending on solvent. Despite structural differences with CHL (absence of three carboxyl groups, presence of lipophilic phytol moiety) and vastly different water solubility, natural chlorophylls are able to form a comparably stable 2:1 complex with DBP in vitro. Chlorophyll a was shown by Dashwood et al (Dashwood et al., 1996) in a similar experiment to complex only very weakly with several heterocyclic amine mutagens. Prior to this, weak binding of Chl to the carcinogenic heterocyclic amine Trp-p-2 (3-amino-1-methyl-5H-pyrido[4,3-b]indole) was reported by Negishi et al (Negishi et al., 1990). Further studies are needed to determine if complex formation in biologically compatible solvents will be a useful indicator of Chl blocking activity in vivo.
Conclusions
This study demonstrated a significant and substantial chemopreventive effect of natural chlorophyll against liver and stomach carcinogenesis in trout when given by dietary co-exposure with carcinogen. Protection was comparable to that shown by CHL, and occurred at Chl concentrations well within the range found in spinach. Chl and CHL were near equally capable of complexing strongly with DBP in vitro, and of reducing systemic bioavailability of DBP to the liver in the in vivo trout co-gavage model as they do in the rat (Simonich et al., 2007). These findings, along with our recently published study in rats (Simonich et al., 2007) provide the first demonstration in any animal models of cancer chemoprotection by dietary natural chlorophyll, which may be a less problematic choice for human intervention than its derivative chlorophyllin.
Acknowledgments
We especially thank Eric Johnson, Greg Gonnerman, and Sheila Cleveland of the Sinnhuber Aquatic Research Laboratory for their excellence in fish rearing, necropsy, and histology. We also thank Dr. Ajoy Velayudin for initial work on Chl purification using CCC. Partly supported through NIH grants CA90890, ES00210, ES03850
Abbreviations
- Chl
chlorophyll
- CHL
chlorophyllin
- DBP
dibenzo[a,l]pyrene
- ESR
electron spin resonance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bailey GS, Williams DE, Hendricks JD. Fish models for environmental carcinogenesis: the rainbow trout. Environ Health Perspect. 1996;104(Suppl 1):5–21. doi: 10.1289/ehp.96104s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum CA, Xu M, Orner GA, Dario Diaz G, Li Q, Dashwood WM, Bailey GS, Dashwood RH. Promotion versus suppression of rat colon carcinogenesis by chlorophyllin and chlorophyll: modulation of apoptosis, cell proliferation, and beta-catenin/Tcf signaling. Mutat Res. 2003:523–524. 217–223. doi: 10.1016/s0027-5107(02)00338-x. [DOI] [PubMed] [Google Scholar]
- Breinholt V, Arbogast D, Loveland P, Pereira C, Dashwood R, Hendricks J, Bailey G. Chlorophyllin chemoprevention in trout initiated by aflatoxin B(1) bath treatment: An evaluation of reduced bioavailability vs. target organ protective mechanisms. Toxicol Appl Pharmacol. 1999;158:141–151. doi: 10.1006/taap.1999.8696. [DOI] [PubMed] [Google Scholar]
- Breinholt V, Hendricks J, Pereira C, Arbogast D, Bailey G. Dietary chlorophyllin is a potent inhibitor of aflatoxin B1 hepatocarcinogenesis in rainbow trout. Cancer Res. 1995a;55:57–62. [PubMed] [Google Scholar]
- Breinholt V, Schimerlik M, Dashwood R, Bailey G. Mechanisms of chlorophyllin anticarcinogenesis against aflatoxin B1: complex formation with the carcinogen. Chem Res Toxicol. 1995b;8:506–514. doi: 10.1021/tx00046a004. [DOI] [PubMed] [Google Scholar]
- Chung WY, Lee JM, Park MY, Yook JI, Kim J, Chung AS, Surh YJ, Park KK. Inhibitory effects of chlorophyllin on 7,12-dimethylbenz[a]anthracene-induced bacterial mutagenesis and mouse skin carcinogenesis. Cancer Lett. 1999;145:57–64. doi: 10.1016/s0304-3835(99)00229-3. [DOI] [PubMed] [Google Scholar]
- Dashwood R, Negishi T, Hayatsu H, Breinholt V, Hendricks J, Bailey G. Chemopreventive properties of chlorophylls towards aflatoxin B1: a review of the antimutagenicity and anticarcinogenicity data in rainbow trout. Mutat Res. 1998;399:245–253. doi: 10.1016/s0027-5107(97)00259-5. [DOI] [PubMed] [Google Scholar]
- Dashwood R, Yamane S, Larsen R. Study of the forces of stabilizing complexes between chlorophylls and heterocyclic amine mutagens. Environ Mol Mutagen. 1996;27:211–218. doi: 10.1002/(SICI)1098-2280(1996)27:3<211::AID-EM6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Dashwood RH. The importance of using pure chemicals in (anti) mutagenicity studies: chlorophyllin as a case in point. Mutat Res. 1997;381:283–286. doi: 10.1016/s0027-5107(97)00221-2. [DOI] [PubMed] [Google Scholar]
- Dashwood RH, Breinholt V, Bailey GS. Chemopreventive properties of chlorophyllin: inhibition of aflatoxin B1 (AFB1)-DNA binding in vivo and anti-mutagenic activity against AFB1 and two heterocyclic amines in the Salmonella mutagenicity assay. Carcinogenesis. 1991;12:939–942. doi: 10.1093/carcin/12.5.939. [DOI] [PubMed] [Google Scholar]
- de Vogel J, Jonker-Termont DS, van Lieshout EM, Katan MB, van der Meer R. Green vegetables, red meat and colon cancer: chlorophyll prevents the cytotoxic and hyperproliferative effects of haem in rat colon. Carcinogenesis. 2005;26:387–393. doi: 10.1093/carcin/bgh331. [DOI] [PubMed] [Google Scholar]
- Dingley KH, Ubick EA, Chiarappa-Zucca ML, Nowell S, Abel S, Ebeler SE, Mitchell AE, Burns SA, Steinberg FM, Clifford AJ. Effect of dietary constituents with chemopreventive potential on adduct formation of a low dose of the heterocyclic amines PhIP and IQ and phase II hepatic enzymes. Nutr Cancer. 2003;46:212–221. doi: 10.1207/S15327914NC4602_15. [DOI] [PubMed] [Google Scholar]
- Dragsted LO, Strube M, Larsen JC. Cancer-protective factors in fruits and vegetables: biochemical and biological background. Pharmacol Toxicol. 1993;72(Suppl 1):116–135. doi: 10.1111/j.1600-0773.1993.tb01679.x. [DOI] [PubMed] [Google Scholar]
- Egner PA, Wang JB, Zhu YR, Zhang BC, Wu Y, Zhang QN, Qian GS, Kuang SY, Gange SJ, Jacobson LP, Helzlsouer KJ, Bailey GS, Groopman JD, Kensler TW. Chlorophyllin intervention reduces aflatoxin-DNA adducts in individuals at high risk for liver cancer. Proc Natl Acad Sci U S A. 2001;98:14601–14606. doi: 10.1073/pnas.251536898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey JW, Stephenson KK, Dinkova-Kostova AT, Egner PA, Kensler TW, Talalay P. Chlorophyll, chlorophyllin and related tetrapyrroles are significant inducers of mammalian phase 2 cytoprotective genes. Carcinogenesis. 2005;26:1247–1255. doi: 10.1093/carcin/bgi068. [DOI] [PubMed] [Google Scholar]
- Ferruzzi MG, Failla ML, Schwartz SJ. Sodium copper chlorophyllin: in vitro digestive stability and accumulation by Caco-2 human intestinal cells. J Agric Food Chem. 2002;50:2173–2179. doi: 10.1021/jf010869g. [DOI] [PubMed] [Google Scholar]
- Guo D, Horio DT, Grove JS, Dashwood RH. Inhibition by chlorophyllin of 2-amino-3-methylimidazo-[4,5-f]quinoline-induced tumorigenesis in the male F344 rat. Cancer Lett. 1995a;95:161–165. doi: 10.1016/0304-3835(95)03882-w. [DOI] [PubMed] [Google Scholar]
- Guo D, Schut HA, Davis CD, Snyderwine EG, Bailey GS, Dashwood RH. Protection by chlorophyllin and indole-3-carbinol against 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-induced DNA adducts and colonic aberrant crypts in the F344 rat. Carcinogenesis. 1995b;16:2931–2937. doi: 10.1093/carcin/16.12.2931. [DOI] [PubMed] [Google Scholar]
- Harttig U, Bailey GS. Chemoprotection by natural chlorophylls in vivo: inhibition of dibenzo[a,l]pyrene-DNA adducts in rainbow trout liver. Carcinogenesis. 1998;19:1323–1326. doi: 10.1093/carcin/19.7.1323. [DOI] [PubMed] [Google Scholar]
- Hasegawa R, Hirose M, Kato T, Hagiwara A, Boonyaphiphat P, Nagao M, Ito N, Shirai T. Inhibitory effect of chlorophyllin on PhIP-induced mammary carcinogenesis in female F344 rats. Carcinogenesis. 1995;16:2243–2246. doi: 10.1093/carcin/16.9.2243. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Schimerlik M, Bailey G. Mechanisms of chlorophyllin anticarcinogenesis: dose-responsive inhibition of aflatoxin uptake and biodistribution following oral co-administration in rainbow trout. Toxicol Appl Pharmacol. 1999;158:132–140. doi: 10.1006/taap.1999.8695. [DOI] [PubMed] [Google Scholar]
- Hayatsu H, Arimoto S, Negishi T. Dietary inhibitors of mutagenesis and carcinogenesis. Mutat Res. 1988;202:429–446. doi: 10.1016/0027-5107(88)90204-7. [DOI] [PubMed] [Google Scholar]
- Hendricks JD, Meyers TR, Shelton DW. Histological progression of hepatic neoplasia in rainbow trout (Salmo gairdneri) Natl Cancer Inst Monogr. 1984;65:321–336. [PubMed] [Google Scholar]
- Hendricks JD, Shelton DW, Loveland PM, Pereira CB, Bailey GS. Carcinogenicity of dietary dimethylnitrosomorpholine, N-methyl-N’-nitro-N-nitrosoguanidine, and dibromoethane in rainbow trout. Toxicol Pathol. 1995;23:447–457. doi: 10.1177/019262339502300402. [DOI] [PubMed] [Google Scholar]
- Hidaka S, Matsumoto E, Toda F. Photoinduced reduction of methylviologen by ascorbate using chlorophyllin in a liposome system. Bull Chem Soc Japan. 1985;58:2007–2010. [Google Scholar]
- Jubert C, Bailey G. Isolation of chlorophylls a and b from spinach by counter-current chromatography. J Chromatogr A. 2007;1140:95–100. doi: 10.1016/j.chroma.2006.11.063. [DOI] [PubMed] [Google Scholar]
- Kenward MG, Roger JH. Small sample inference for fixed effects from restricted maximum likelihood. Biometrics. 1997;53:983–997. [PubMed] [Google Scholar]
- Kim J, Yook JI, Park KK, Jung SY, Hong JC, Kim KJ, Kim JA, Chung WY. Anti-promotion effect of chlorophyllin in DMBA-TPA-induced mouse skin carcinogenesis. Anticancer Res. 2000;20:1493–1498. [PubMed] [Google Scholar]
- Kohlmeier L, Simonsen N, Mottus K. Dietary modifiers of carcinogenesis. Environ Health Perspect. 1995;103(Suppl 8):177–184. doi: 10.1289/ehp.95103s8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BC, Hendrick JD, Bailey GS. In: Mycotoxins and Animal Feedingstuff: Natural Occurrence, Toxicity and Control. Smith JE, editor. CRC Press; Boca Raton, FL: 1991. pp. 607–626. [Google Scholar]
- Negishi T, Arimoto S, Nishizaki C, Hayatsu H. Inhibition of the genotoxicity of 3-amino-1-methyl-5H-pyrido[4,3-b]indole (Trp-P-2) in Drosophila by chlorophyll. Basic Life Sci. 1990;52:341–344. doi: 10.1007/978-1-4615-9561-8_29. [DOI] [PubMed] [Google Scholar]
- Negishi T, Nakano H, Kitamura A, Itome C, Shiotani T, Hayatsu H. Inhibitory activity of chlorophyllin on the genotoxicity of carcinogens in Drosophila. Cancer Lett. 1994;83:157–164. doi: 10.1016/0304-3835(94)90313-1. [DOI] [PubMed] [Google Scholar]
- Negishi T, Rai H, Hayatsu H. Antigenotoxic activity of natural chlorophylls. Mutat Res. 1997;376:97–100. doi: 10.1016/s0027-5107(97)00030-4. [DOI] [PubMed] [Google Scholar]
- Nelson RL. Chlorophyllin, an antimutagen, acts as a tumor promoter in the rat-dimethylhydrazine colon carcinogenesis model. Anticancer Res. 1992;12:737–739. [PubMed] [Google Scholar]
- Ong TM, Whong WZ, Stewart J, Brockman HE. Chlorophyllin: a potent antimutagen against environmental and dietary complex mixtures. Mutat Res. 1986;173:111–115. doi: 10.1016/0165-7992(86)90086-2. [DOI] [PubMed] [Google Scholar]
- Park KK, Surh YJ. Chemopreventive activity of chlorophyllin against mouse skin carcinogenesis by benzo[a]pyrene and benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide. Cancer Lett. 1996;102:143–149. doi: 10.1016/0304-3835(96)04173-0. [DOI] [PubMed] [Google Scholar]
- Pratt MM, Reddy AP, Hendricks JD, Pereira C, Kensler TW, Bailey GS. The importance of carcinogen dose in chemoprevention studies: quantitative interrelationships between, dibenzo[a,l]pyrene dose, chlorophyllin dose, target organ DNA adduct biomarkers, and final tumor outcome. Carcinogenesis. 2006 doi: 10.1093/carcin/bgl174. Epub ahead of print, Sept 14. [DOI] [PubMed] [Google Scholar]
- Reddy AP, Harttig U, Barth MC, Baird WM, Schimerlik M, Hendricks JD, Bailey GS. Inhibition of dibenzo[a,l]pyrene-induced multi-organ carcinogenesis by dietary chlorophyllin in rainbow trout. Carcinogenesis. 1999;20:1919–1926. doi: 10.1093/carcin/20.10.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romert L, Curvall M, Jenssen D. Chlorophyllin is both a positive and negative modifier of mutagenicity. Mutagenesis. 1992;7:349–355. doi: 10.1093/mutage/7.5.349. [DOI] [PubMed] [Google Scholar]
- Sato M, Konagai K, Kuwana T, Kimura R, Murata T. Effect of sodium copper chlorophyllin on lipid peroxidation. VII. Effect of its administration on the stability of rat liver lysosomes. Chem Pharm Bull (Tokyo) 1984;32:2855–2858. doi: 10.1248/cpb.32.2855. [DOI] [PubMed] [Google Scholar]
- Sesink AL, Termont DS, Kleibeuker JH, Van der Meer R. Red meat and colon cancer: the cytotoxic and hyperproliferative effects of dietary heme. Cancer Res. 1999;59:5704–5709. [PubMed] [Google Scholar]
- Simonich MT, Egner PA, Roebuck BD, Orner GA, Jubert C, Pereira C, Groopman JD, Kensler TW, Dashwood RH, Williams DE, Bailey GS. Natural chlorophyll inhibits aflatoxin B1-induced multi-organ carcinogenesis in the rat. Carcinogenesis. 2007;28:1294–1302. doi: 10.1093/carcin/bgm027. [DOI] [PubMed] [Google Scholar]
- Sinnhuber RO, Hendricks JD, Wales JH, Putnam GB. Neoplasms in rainbow trout, a sensitive animal model for environmental carcinogenesis. Ann N Y Acad Sci. 1978;298:389–408. doi: 10.1111/j.1749-6632.1977.tb19280.x. [DOI] [PubMed] [Google Scholar]
- Tachino N, Guo D, Dashwood WM, Yamane S, Larsen R, Dashwood R. Mechanisms of the in vitro antimutagenic action of chlorophyllin against benzo[a]pyrene: studies of enzyme inhibition, molecular complex formation and degradation of the ultimate carcinogen. Mutat Res. 1994;308:191–203. doi: 10.1016/0027-5107(94)90154-6. [DOI] [PubMed] [Google Scholar]
- Warner JR, Nath J, Ong TM. Antimutagenicity studies of chlorophyllin using the Salmonella arabinose-resistant assay system. Mutat Res. 1991;262:25–30. doi: 10.1016/0165-7992(91)90101-9. [DOI] [PubMed] [Google Scholar]
- Wattenberg LW. Inhibition of carcinogenesis by naturally-occurring and synthetic compounds. Basic Life Sci. 1990;52:155–166. doi: 10.1007/978-1-4615-9561-8_12. [DOI] [PubMed] [Google Scholar]
- Whong WZ, Stewart J, Brockman HE, Ong TM. Comparative antimutagenicity of chlorophyllin and five other agents against aflatoxin B1-induced reversion in Salmonella typhimurium strain TA98. Teratog Carcinog Mutagen. 1988;8:215–224. doi: 10.1002/tcm.1770080405. [DOI] [PubMed] [Google Scholar]
- Williams DE, Bailey GS, Reddy A, Hendricks JD, Oganesian A, Orner GA, Pereira CB, Swenberg JA. The rainbow trout (Oncorhynchus mykiss) tumor model: recent applications in low-dose exposures to tumor initiators and promoters. Toxicol Pathol. 2003;(31 Suppl):58–61. [PubMed] [Google Scholar]
- Wu ZL, Chen JK, Ong T, Brockman HE, Whong WZ. Antitransforming activity of chlorophyllin against selected carcinogens and complex mixtures. Teratog Carcinog Mutagen. 1994;14:75–81. doi: 10.1002/tcm.1770140204. [DOI] [PubMed] [Google Scholar]
- Xu M, Orner GA, Bailey GS, Stoner GD, Horio DT, Dashwood RH. Post-initiation effects of chlorophyllin and indole-3-carbinol in rats given 1,2-dimethylhydrazine or 2-amino-3-methyl- imidazo. Carcinogenesis. 2001;22:309–314. doi: 10.1093/carcin/22.2.309. [DOI] [PubMed] [Google Scholar]
- Yun CH, Jeong HG, Jhoun JW, Guengerich FP. Non-specific inhibition of cytochrome P450 activities by chlorophyllin in human and rat liver microsomes. Carcinogenesis. 1995;16:1437–1440. doi: 10.1093/carcin/16.6.1437. [DOI] [PubMed] [Google Scholar]