

Abstract
The genetic basis of spontaneous melanoma formation in spotted dorsal (Sd) Xiphophorus platyfish–swordtail hybrids has been studied for decades, and is adequately explained by a two-gene inheritance model involving a sex-linked oncogene, Xmrk, and an autosomal tumor suppressor, DIFF. The Xmrk oncogene encodes a receptor tyrosine kinase related to EGFR; the nature of the DIFF tumor suppressor gene is unknown. We analyzed the genetic basis of UV-B-induced melanoma formation in closely related, spotted side platyfish–swordtail hybrids, which carry a different sex-linked pigment pattern locus, Sp. We UV-irradiated spotted side Xiphophorus platyfish–swordtail backcross hybrids to induce melanomas at frequencies 6-fold higher than occur spontaneously in unirradiated control animals. To identify genetic determinants of melanoma susceptibility in this UV-inducible Xiphophorus model, we genotyped individual animals from control and UV-irradiated experimental regimes using allozyme and DNA restriction fragment length polymorphisms and tested for joint segregation of genetic markers with pigmentation phenotype and UV-induced melanoma formation. Joint segregation results show linkage of a CDKN2-like DNA polymorphism with UV-B-induced melanoma formation in these hybrids. The CDKN2-like polymorphism maps to Xiphophorus linkage group V and exhibits recombination fractions with ES1 and MDH2 allozyme markers consistent with previous localization of the DIFF tumor suppressor locus. Our results indicate that the CDKN2-like sequence we have cloned and mapped is a candidate for the DIFF tumor suppressor gene.
Keywords: CDKN2, Xiphophorus hybrid melanoma, DIFF tumor suppressor gene, UV
Genetic hybrids between species of the genus Xiphophorus (Teleostei: Poeciliidae) exhibit spontaneous melanoma formation in several different cross types and have been used for decades to investigate genetic factors contributing to melanoma formation (1). The most studied and best understood Xiphophorus hybrid melanoma is the spotted dorsal Gordon–Kosswig platyfish–swordtail model (2–5), represented by genetic hybrids derived from crossing F1 hybrids between the platyfish Xiphophorus maculatus Jp 163 A and the swordtail Xiphophorus helleri back to X. helleri. Melanoma formation in this tumor model is genetically controlled by inheritance of a sex-linked receptor tyrosine kinase gene (Xmrk), associated with the spotted dorsal (Sd) pigment pattern locus, and segregation of an autosomal locus in Xiphophorus linkage group (LG) V, variously referred to in the literature as DIFF, RDIFF, and R (3–6). The DIFF locus is believed to regulate macromelanophore pigment cell differentiation (4, 6), and behaves in the Gordon–Kosswig model as a classical tumor suppressor for which loss of species-specific alleles in pigmented backcross hybrids results in melanoma formation according to simple, Mendelian inheritance (1, 3–5).
Elegant experiments have shown that Xmrk is a duplicated gene, which has adventitiously acquired the promoter from another gene (7, 8). It has been postulated that expression of the oncogenic Xmrk gene duplicate is regulated by the DIFF autosomal locus (5, 8). Supporting this hypothesis are studies showing Xmrk overexpression (9), and differential expression (10) of the oncogenic Xmrk and its protooncogene copy [referred to as INV-Xmrk (11) or Xmrk-1 (10)], in spontaneous melanomas. Furthermore, the level of Xmrk overexpression correlates with the degree of malignancy of melanomas (7), and Xmrk overexpression in transgenic medaka (Oryzias latipes) is tumorigenic (12). These results strongly suggest that the regulation of Xmrk gene expression is a critical determinant of carcinogenesis in these tumor models. However, no candidate sequence for DIFF has been identified, and its putative role in regulating Xmrk expression has not been directly shown, but is inferred from genetic studies.
Genetic linkage analyses of melanoma in humans have indicated that alterations in the CDKN2 gene, encoding the p16 protein, explains part of the clinical phenotype of familial atypical multiple-mole melanoma (FAMMM) syndrome (13, 14). CDKN2 is also altered in many other primary tumors (15). Germ-line mutations in CDKN2 have been detected in which p16 dysfunction results in the inability to bind cdk4 in vitro, and implicate CDKN2 in development of heritable melanoma (16). Moreover, there is correlation of loss of expression of CDKN2 with the invasive stage of melanoma progression (17). There is also evidence for UV induction of CDKN2 mutations in human melanoma cell lines derived from patients without a family history of melanomas (18). Thus, CDKN2 is a strong candidate for a human melanoma susceptibility gene.
UV-inducible Xiphophorus hybrid melanoma models have been developed to investigate the potential role of excessive sunlight exposure in melanoma formation (19, 20). Setlow et al. (19) used backcross hybrids from a spotted side (Sp) platyfish–swordtail hybrid model, closely related to the classical, spotted dorsal (Sd) Gordon–Kosswig spontaneous melanoma model, to demonstrate that UV irradiation was effective for melanoma induction, resulting in melanoma incidences of up to 40% in UV-B-irradiated backcross hybrids. However, the genetic basis of the UV-inducible, spotted side platyfish–swordtail hybrid model was not investigated in this initial study. We report results here that (i) establish that a probable homologue of a mammalian CDKN2 is located in Xiphophorus LG V at or near the location of the DIFF tumor suppressor locus, and (ii) demonstrate that CDKN2-like genotypes are strongly correlated with susceptibility to UV-induced melanoma formation in the spotted side Xiphophorus hybrid model.
MATERIALS AND METHODS
Animal Strains and Genetic Hybridization.
Parental and hybrid fish were derived from inbred genetic stocks maintained at the Xiphophorus Genetic Stock Center (Southwest Texas State University, San Marcos, TX). Stocks of platyfish strains X. maculatus Jp 163 A and Jp 163 B have been maintained by brother–sister inbreeding since establishment of X. maculatus Jp 163 from collections in the Rio Jamapa (Veracruz, Mexico), in 1939. These two platyfish strains are descended from the same X. maculatus female, and differ only in that different pigment pattern loci are carried on the X chromosomes, spotted dorsal (Sd) in the case of Jp 163 A, and spotted side (Sp) in the case of Jp 163 B (21). Stocks of the swordtail X. helleri Sarabia strain have been maintained in closed colony since establishment from collections in the Rio Sarabia (Oaxaca, Mexico), in 1963. Interspecific F1 hybrids between X. maculatus Jp 163 B and X. helleri were produced by artificial insemination (22). Matings established between F1 hybrids and X. helleri parental fish resulted in production of backcross hybrids, each of which exhibits heterozygosity for X. maculatus and X. helleri alleles, or homozygosity for X. helleri alleles, at one-half of genetic loci as determined by independent assortment of chromosomes at meiosis.
PCR Amplification and Cloning of a CDKN2-Like Sequence from X. maculatus.
DNA was isolated from brain harvested from X. maculatus Jp 163 A according to a previously published protocol (23). Amplification primers were designed based on inspection of human and murine exon 2 CDKN2A (p16) and CDKN2B (p15) sequences (24–26). “Touchdown” PCR methodology, as described by Don et al. (27) and Roux (28), was used to amplify the Xiphophorus CDKN2-like sequence. Forward and reverse primers used for amplication were, respectively, P16F1: (GTCATGATGATGGGC) and P16R2: (GCGTGTCCAGGAAGC). The 130-bp amplification product was cloned into the pCRII plasmid by TA cloning (TA cloning kit, Invitrogen). Dideoxy sequencing (29) and Vent polymerase cycle sequencing (Circumvent kit, NEB, Beverly, MA) methods were used to determine the nucleotide sequence of the cloned insert.
UV-Irradiation of Backcross Hybrids.
Backcross hybrids were irradiated in a UV-B exposure protocol based on the data of Setlow et al. (19). Irradiation conditions used cellulose acetate filtration of FS-20 sunlamps, with a cut-off of wavelengths below 290 nm, and mimic the solar spectrum. Five days after birth, individual broods of 6–24 animals were irradiated from above with three Westinghouse FS-20 sunlamps filtered through cellulose acetate film (Kodacel, Eastman-Kodak) in 2.5-gallon aquaria containing water to a depth of 5 cm. The dose rate was adjusted using a rheostat to modulate sunlamp intensity, resulting in a rate of 0.33 J/m2 per sec, measured through the cellulose acetate film. Fluence was measured using a Model 1L 1400 A Radiometer/Photometer with a UVB-1 probe (International Light, Newburyport, MA). A total fluence of 1500 J/m2 (incident to the surface of 5 cm of water) was delivered in equal doses of 300 J/m2 per day for 5 days. Care was taken to keep irradiated fish completely dark for at least 16 hr after irradiation and in subdued light over the course of the total 5-day irradiation protocol. As they matured, Sp-inheriting fish were scored for macromelanophore spotting phenotype (light or heavy) and for presumptive tumors at 4 and 6 months, then sacrificed at 6 months for tissues to perform isozyme and DNA analyses to determine inheritance of genetic markers. Tumors were excised from the animals at the time of sacrifice and fixed in buffered 10% formalin.
Genetic Linkage Analysis.
Dissection and tissue preparation for starch gel electrophoresis of proteins followed described methods (30). A listing of polymorphic proteins analyzed in this study, with conditions for electrophoresis and histochemical staining, may be found in Morizot and Schmidt (31). Tissues for DNA extraction (testis, spleen, kidney, gill) were dissected as rapidly as possible and flash frozen in a dry ice-ethanol bath, then stored at −80°C prior to DNA isolation. Genomic DNA for restriction fragment length polymorphism (RFLP) analysis was prepared from tissues of backcross hybrids using commercially available DNA extraction kits (either PureGene Kit, Gentra Systems or IsoQuick DNA Isolation Kit, MicroProbe, Garden Grove, CA). Purified DNA was dissolved in 0.1 × TE (1 mM Tris·HCl/0.1 mM EDTA, pH 7.5) and stored at −20°C until use. DNA samples were digested with appropriate restriction endonucleases, electrophoresed in 0.7% agarose gels, and blotted to Magna nylon membranes (Micron Separations) by capillary transfer. Conditions of hybridization and washing were essentially as described (32–34), except that in some cases random prime labeling (DecaPrime II Kit, Ambion, Austin, TX) instead of nick translation was used to isotopically label DNA probes. DNA probes used to map Xiphophorus loci defined by RFLPs were excised as inserts from plasmids and purified from agarose gels prior to use in Southern hybridization. The origin of each DNA probe (except ACTBL1 and CDKN2) and its use in genetic mapping in Xiphophorus genetic hybrids have been reported (32–35). The ACTBL1 polymorphism was detected with a Xiphophorus β-actin cDNA (R.B.W., unpublished work).
DNA RFLP and isozyme nomenclature generally follows standardized human gene nomenclature (36); pigment pattern gene symbols are described in Morizot et al. (37). Allozyme and DNA RFLP phenotypes were scored as heterozygotes or homozygotes in accordance with codominant inheritance expectations. Segregation and linkage analyses of the resulting genotypic data were performed with the computer programs mapmanager 2.6.5 (available from Kenneth F. Manly, Roswell Park Memorial Institute, Buffalo, NY) to generate recombination values, and mapmaker 3.0 (38) to calculate maximum likelihood gene map orders and map interval information.
Histological Evaluation of Melanomas.
Tumors preserved in buffered formalin were imbedded in paraffin blocks, cut into 6-μm sections, and stained with hematoxylin/eosin. Samples were shipped to Avril Woodhead (Brookhaven National Laboratory, Upton, NY), who confirmed the characteristics of invasive melanomas using criteria established in the original description of the spotted side hybrid melanoma model (19). These included intense proliferation of dermal macromelanophores accompanied by inflammation at the margins of the tumors, and an ulcerated, swirled mass of spindle-shaped melanocytes in the interior of the malignant melanotic nodules, with invasion into surrounding tissues.
RESULTS
Joint Segregation Analysis of the CDKN2-Like Polymorphism in Backcross Hybrids.
The nucleotide and computer-translated amino acid sequences for the PCR amplification product obtained using the CDKN2 amplimers described in Materials and Methods are shown in Fig. 1. As shown by the comparisons of Fig. 1B, the Xiphophorus sequence in this region is conserved at the amino acid level with human CDKN2A (p16), CDKN2B (p15), and CDKN2D (p19) sequences (24–26, 39), and is only distantly related to human ankyrin (40) or to the notch homologue from goldfish (GenBank accession no. U09191U09191); comparisons with murine CDKN2 family members (not shown) also suggest that the Xiphophorus CDKN2-like sequence corresponds to a CDKN2-related gene.
Figure 1.

Xiphophorus CDKN2-like nucleotide sequence and computer-translated amino acid sequences of ankyrin-related genes. (A) Xiphophorus nucleotide sequence with amplimers underlined. (B) Computer-generated translations of nucleotide sequences from Xiphophorus, human, and goldfish ankyrin-related sequences, as indicated.
Fig. 2 is a representative Southern blot of PstI-digested DNA from backcross hybrids of the X. helleri × (X. maculatus Jp 163 B × X. helleri) cross type. A distinct RFLP between X. maculatus (≈5-kb band) and X. helleri (≈1-kb band) CDKN2-hybridizing DNA is observable. This polymorphism was designated CDKN2L1 based on homology with mammalian CDKN2 sequences (Fig. 1). Table 1 shows the results of joint segregation analysis of CDKN2L1 genotypes tested against 37 informative polymorphic markers available in this cross. These data clearly establish linkage of the CDKN2L1 sequence with two allozyme (ES1 and MDH2) and one RFLP (ACTBL1) markers in LG V. Multipoint linkage analysis indicates that location of CDKN2L1 between ES1 and MDH2 is > 2500 times more likely than alternate locations: a gene order of MDH2–CDKN2L1–ES1–ACTBL1 minimizes multiple crossovers in the subset of individuals informative for all LG V loci (totals of 28 single crossovers, 6 double crossovers, and 2 triple crossovers). Fig. 3 illustrates this gene order, and indicates recombination fractions observed between markers.
Figure 2.
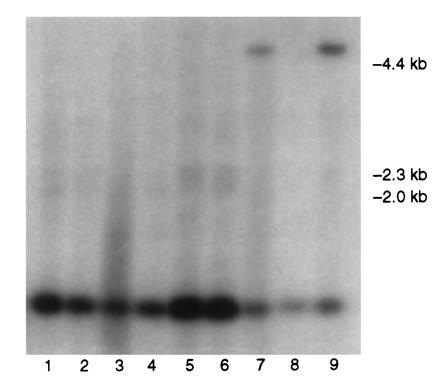
Representative Southern blot of CDKN2L1 PstI polymorphism in backcross hybrids. Lanes 1–6 and 8, homozygotes; lanes 7 and 9, heterozygotes.
Table 1.
Joint segregation analysis of CDKN2-like polymorphism with 37 allozyme, RFLP, and pigment pattern loci informative in X. helleri × (X. maculatus Jp 163 B × X. helleri) backcross hybrids
| Locus* | LG | No. of parentals | No. of recombinants | r† | χ2 |
|---|---|---|---|---|---|
| ACO1 | XIV | 57 | 45 | 0.44 | 1.4 |
| ACTBL1 | V | 65 | 19 | 0.23 | 25.2‡ |
| ADA | I | 46 | 44 | 0.49 | 0 |
| ATP | III | 27 | 44 | 0.62 | 4.1 |
| CKM | XI | 42 | 44 | 0.51 | 0.1 |
| EGFR | VI | 16 | 15 | 0.48 | 0 |
| ES1 | V | 77 | 23 | 0.23 | 29.2‡ |
| ES2 | II | 58 | 53 | 0.48 | 0.2 |
| ES3 | II | 41 | 32 | 0.44 | 1.1 |
| ES5 | II | 18 | 18 | 0.50 | 0 |
| ES7 | III | 9 | 10 | 0.50 | 0.1 |
| FYN | XV | 32 | 20 | 0.39 | 2.8 |
| GALT1 | VIII | 50 | 42 | 0.47 | 0.7 |
| GAPD1 | III | 40 | 60 | 0.60 | 2.0 |
| GDA | XII | 30 | 40 | 0.57 | 1.4 |
| GLA | XV | 52 | 47 | 0.48 | 0.3 |
| G6PD | I | 60 | 42 | 0.41 | 4.8 |
| GPI1 | IV | 25 | 25 | 0.50 | 0 |
| IDH1 | IV | 33 | 32 | 0.50 | 0 |
| IDH2 | VII | 44 | 52 | 0.54 | 0.7 |
| ITP | UA | 25 | 19 | 0.43 | 0.8 |
| LIG1 | VI | 40 | 48 | 0.49 | 0.8 |
| MACR | XXIV | 39 | 63 | 0.62 | 5.7 |
| MDH2 | V | 70 | 17 | 0.20 | 32.3‡ |
| MPI | II | 48 | 51 | 0.52 | 0.1 |
| PEPS | XII | 45 | 37 | 0.45 | 0.8 |
| PGAM1 | XI | 55 | 39 | 0.42 | 2.7 |
| PGAM2 | VIII | 42 | 48 | 0.53 | 0.4 |
| PGD | I | 46 | 45 | 0.50 | 0 |
| PGK | XI | 50 | 41 | 0.45 | 0.9 |
| PGM | IX | 49 | 50 | 0.50 | 0 |
| PK1 | IV | 24 | 26 | 0.52 | 0.1 |
| PVALB2 | X | 38 | 46 | 0.55 | 0.8 |
| SRC | I | 27 | 24 | 0.47 | 0.2 |
| TP53 | XIV | 28 | 21 | 0.43 | 1.0 |
| UMPK | VI | 47 | 52 | 0.53 | 0.3 |
| YES | VI | 47 | 45 | 0.49 | 0 |
Figure 3.
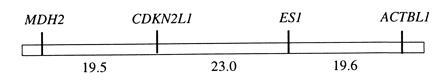
Genetic linkage markers in Xiphophorus LG V. Numerical values refer to percent recombination between markers (see text).
UV-Induced Melanoma Formation in Hybrids.
The general phenotypes of parental and hybrid fish are shown in Fig. 4. The macromelanophore spotting of the spotted side pigmentation pattern in parental X. maculatus Jp 163 B animals (Fig. 4B) is overexpressed in F1 hybrids (Fig. 4C), resulting in hyperplasia and a “marbled” pigmentation (19). Pigmented backcross hybrids inheriting the Sp pigment pattern locus constituted approximately one-half of the total backcross progeny. Pigmented backcross hybrids fell into two groups of roughly one-half heavily pigmented (Hp) fish (Fig. 4E) and one-half with a light pigmentation (Lp) pattern (Fig. 4D) resembling the F1 hybrid pigmentation phenotype (Fig. 4C), although with somewhat more extensive hyperplasia. Determination of Lp or Hp phenotypes was not possible until the fish had matured to about 2 months of age; by 4 months of age, these pigmentation phenotypes were clearly discernible and distinct.
Figure 4.
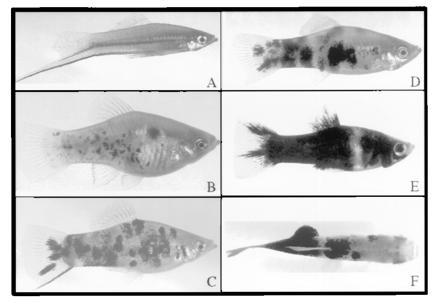
Parental and F1 hybrid fish from the X. helleri × (X. maculatus Jp 163 B × X. helleri) cross type. (A) X. helleri, Sarabia strain, male parent; (B) X. maculatus Jp 163 B, female parent; (C) F1 hybrid; (D) light pigmentation (Lp) phenotype backcross hybrid; (E) heavy pigmentation (Hp) phenotype backcross hybrid; (F) tumor-bearing, UV-irradiated backcross hybrid. Animals shown in D and E are from the same brood, photographed at approximately 5 months of age.
Data presented in Table 2 show the frequencies of histologically confirmed melanomas in UV-B-irradiated and unirradiated control animals. An example of a fish with such a tumor is shown in Fig. 4F. The results in Table 2 show that the spontaneous incidence of melanomas at 4 and 6 months was very low, with no tumors detected in lightly pigmented hybrids, and only one histologically confirmed melanoma in heavily pigmented hybrids. In UV-irradiated backcross hybrids, lightly pigmented animals exhibited a low incidence of melanoma formation (5.4%), but the incidence of melanomas formed in heavily pigmented hybrids was 34.8%, or more than 6 times the incidence in unirradiated, heavily pigmented hybrids (5.5%). These results independently confirm the findings of Setlow et al. (19), demonstrating that Sp pigment pattern-derived platyfish–swordtail backcross hybrids are susceptible to UV-induced melanoma formation.
Table 2.
Effects of UV-B irradiation on melanoma formation in X. helleri × (X. maculatus Jp 163 B × X. helleri) backcross hybrids
| Scoring | Unirradiated
|
UV-B
irradiated
|
||||||
|---|---|---|---|---|---|---|---|---|
| Light phenotype
|
Heavy
phenotype
|
Light phenotype
|
Heavy phenotype
|
|||||
| Tumor | No tumor | Tumor | No tumor | Tumor | No tumor | Tumor | No tumor | |
| 4 months | 0 | 27 | 1 | 17 | 1 | 56 | 16 | 30 |
| 6 months | 0 | 27 | 1 | 17 | 3 | 53 | 16 | 30 |
Linkage of CDKN2L1 with UV-Induced Melanoma Formation.
Table 3 presents joint segregation results for CDKN2L1 genotypes with pigmentation phenotypes and UV-induced neoplasms in backcross hybrids. Analysis of the inheritance of Lp and Hp phenotypes represents a genetic test of DIFF regulation of melanocytic hyperplasia in backcross hybrids; the progressive loss of this regulation correlates with segregation of X. maculatus DIFF in Sd, Gordon–Kosswig backcross hybrids, and results in pigment pattern enhancement (1, 3–6). In Sd hybrids, inheritance of both Lp and Hp phenotypes is strongly associated with LG V genotypes, as is also spontaneous melanoma formation from melanoblasts of the spotted dorsal pigment pattern in Hp animals (1, 3–5). Surprisingly, the data in Table 3 show only a weak association of combined Lp and Hp phenotypes with CDKN2L1 in Sp backcross hybrids, and no linkage at all with Lp phenotypes, although linkage of CDKN2L1 with both UV-induced hyperplastic nodular lesions and the subset of histologically confirmed UV-induced melanomas is highly significant. Only the Hp phenotype is significantly associated with CDKN2L1 genotypes; this LG V association is confirmed by linkage of Hp with ES1 (42 parentals, 14 recombinants; χ2 = 14.0). Thus, although there is significant linkage of CDKN2L1 genotypes with UV-induced tumor phenotypes, the LG V DIFF locus does not appear to solely regulate melanocytic hyperplasia in Sp hybrids. This result is in marked contrast to the extensively documented co-inheritance of pigment pattern phenotypes and spontaneous melanoma formation in Sd Gordon–Kosswig platyfish–swordtail hybrids (1–5).
Table 3.
Joint segregation of CDKN2L1 genotypes with pigmentation and tumor phenotypes in X. helleri × (X. maculatus Jp 163 B × X. helleri) backcross hybrids
| Phenotype† | No. of parentals | No. of recombinants | r‡ | χ2 |
|---|---|---|---|---|
| Lp | 7 | 10 | 0.59 | 0.5 |
| Hp | 30 | 11 | 0.27 | 8.8** |
| Lp + Hp | 37 | 21 | 0.36 | 6.1* |
| UV-HNL | 28 | 3 | 0.10 | 20.2** |
| UV-HCM | 17 | 2 | 0.11 | 11.8** |
Lp, light pigmentation phenotype; Hp, heavy pigmentation phenotype; UV-HNL, UV-induced hyperplastic nodular lesions; UV-HCM, UV-induced histologically confirmed melanomas.
Recombination fraction.
, P < 0.05.
, P < 0.01.
, P < 0.001.
DISCUSSION
Melanoma incidence shows an alarming worldwide increase (41–43). Heredity, target cell susceptibility, and excessive sunlight exposure are all believed to be factors in cutaneous malignant melanoma (44, 45); however, a precise role for each of these factors in melanoma formation has not been established (45). CDKN2A, encoding the p16 gene product, is a strong candidate for a human melanoma susceptibility gene, based on pedigree analyses of families with hereditary melanomas (13, 14, 46), and genetic and biochemical studies of both hereditary and sporadic melanomas (16, 17). Mutations in CDKN2A consistent with UV induction have recently been reported in human melanoma cell lines (18), retrospectively suggesting a possible sunlight etiology. Directly testing a role for sunlight exposure in the etiology of melanoma, however, will require experiments in animal models exhibiting sunlight-inducible melanoma formation; the most useful of these models will be amenable to genetic analysis and offer correspondence between human and animal genes implicated in melanoma formation.
Xiphophorus genetic hybrids offer both spontaneous and sunlight-inducible melanoma models. Spontaneous melanoma formation in Gordon–Kosswig platyfish–swordtail hybrids has been studied for decades, and represents an experimental paradigm for hereditary tumor formation (1, 3–5). Recently, work by Setlow and colleagues has established the usefulness of other Xiphophorus hybrids for investigation of sunlight-inducible melanoma formation (19, 20). In this study, we subjected one of these sunlight-inducible Xiphophorus melanoma models, closely related to the classical Gordon–Kosswig hybrid model, to linkage analysis for the inheritance of polymorphic DNA and protein markers with UV-induced melanoma formation. One of these markers is a CDKN2-like DNA sequence we recovered by PCR amplification from the X. maculatus genome. This CDKN2-related sequence maps to Xiphophorus LG V, in the close vicinity of the DIFF locus (Table 1, Fig. 3). Furthermore, there is significant linkage of CDKN2L1 genotypes to UV-induced melanoma formation in Sp platyfish–swordtail backcross hybrids (Table 3). These results, and the evidence from a large number of studies implicating CDKN2A in human melanomas, establish a strong basis for considering this CDKN2-related sequence from Xiphophorus to be a candidate for the DIFF tumor suppressor gene.
The Xiphophorus CDKN2-related DNA sequence we have cloned and mapped exhibits an open reading frame with significant homology to a region in exon 2 of human CDKN2A coding for parts of the second and third of four ankyrin domain repeats (Fig. 1). Human sequences coding for homologous ankyrin domain regions in the p15 (CDKN2B) and p19 (CDKN2D) proteins likewise show extensive amino acid identity with the fish sequence (18 of 33 amino acids are identical compared with human p15 and p16; 18 of 32 compared with human p19). Human p18 possesses five ankyrin repeats and is somewhat less similar (16 of 32 amino acids identical) to the translated Xiphophorus sequence. Numerous possible alignments of non-CDKN2-encoded proteins with ankyrin domains are far less similar to the translated fish sequence than are sequences of CDKN2 gene family members: in Fig. 1, the best alignments produced by clustal w (47) software yield only 10 of 33 identical amino acids for the goldfish notch gene (6 ankyrin repeats) and 9 of 34 identical amino acids for the prototypical human ankyrin gene (24 ankyrin repeats). Comparisons with mouse and rat sequences (data not shown) likewise support assignment of the fish DNA sequence to the CDKN2 gene family, but establishment of orthology with a particular family member is not possible with the available sequence data. In future experiments, it will be very interesting to determine the size of the CDKN2 gene family in Xiphophorus, and to establish orthology and gene map locations for comparison to mammalian CDKN2 loci.
Results of the joint segregation analysis of CDKN2L1 with the 37 other polymorphic genetic markers available in X. helleri × (X. maculatus Jp 163 B × X. helleri) backcross hybrids unequivocally establish its linkage with LG V markers ES1, MDH2, and ACTBL1 (Table 1), and strongly support the gene order shown in Fig. 3. Precise localization of the DIFF tumor suppressor gene in Xiphophorus LG V has proven to be difficult, both in Gordon–Kosswig hybrids and in other Xiphophorus melanoma models, primarily because of variable recombination estimates with other LG V markers. Recombination estimates with ES1 have ranged from ≈10-38% in various Gordon–Kosswig melanoma crosses, in a few cases being higher than estimates with MDH2, which often exceed 30% (48–50). While the reasons for such variability remain unclear, they may indicate interspecific differences in gene arrangement (4), or misclassification of intermediate pigment pattern intensity in some backcross individuals (48), or could reflect variable development of melanomas with different etiologies. Our localization of the CDKN2L1 sequence in LG V is thus of considerable interest in assessment of the likelihood of its identity with DIFF. Our results from the Sp platyfish–swordtail UV-inducible melanoma model suggest that the CDKN2L1 sequence resides near the predicted location of DIFF in LG V midway between ES1 and MDH2 (Fig. 3). Unfortunately, other LG V loci are uninformative in this cross. An additional marker, GLYDH, has been mapped between ES1 and MDH2 in LG V, again with variable recombination in different cross types ranging from 5–29% with MDH2 (37, 51). Recently, we analyzed the inheritance of pigmentation phenotypes and tumor susceptibility in another inducible Xiphophorus melanoma model, in backcross hybrids of the cross type Xiphophorus couchianus × (X. maculatus Jp 163 B × X. couchianus), which is informative for GLYDH (52). Our preliminary linkage analyses (unpublished data) of this hybrid indicate a gene order of GLYDH–CDKN2L1–ACTBL1, confirming the localization of CDKN2L1 in the DIFF region of LG V reported here.
The critical result in this study of significant association of CDKN2L1 genotypes with UV melanoma induction (Table 3) strongly indicates that a CDKN2-related tumor susceptibility gene is a likely candidate for the DIFF tumor suppressor identified in the Gordon–Kosswig spontaneous hybrid melanoma model. However, results of Table 3 indicate that, although CDKN2L1 genotypes are strongly associated with UV-induced tumor formation, there is only a weak association, or none at all, with inheritance of Lp and Hp pigmentation phenotypes in Sp platyfish–swordtail hybrids. In the Gordon–Kosswig model, it is impossible to separate genetic factors controlling pigmentation from those controlling melanoma susceptibility, because all heavily pigmented individuals develop melanomas with age, whereas almost no lightly pigmented individuals develop tumors during the usual time course of experiments (≤1 year). Several other Xiphophorus crosses yield heavy and light phenotypes in Mendelian proportions in backcrosses and variable frequencies of spontaneous melanomas in heavily pigmented individuals, independent of genotype at LG V markers (4, 51). The lack of association of Lp phenotypes with CDKN2L1 and other LG V genetic markers is therefore not without precedent in these hybrid models.
In the Sp platyfish–swordtail hybrid model, loss of X. maculatus alleles in the LG V region containing DIFF does not ineluctably lead to formation of nodular lesions and melanomas in Sp-inheriting backcross hybrids, but does appear to be a necessary precondition for UV-induced tumorigenesis, which occurs only in heavily pigmented backcross hybrids at an appreciable frequency (Table 2). The strong correlation of CDKN2L1 genotypes with tumor susceptibility (Table 3), coupled with its localization to the DIFF region of LG V, suggests that a CDKN2 gene is a likely candidate for the DIFF tumor suppressor gene controlling development of malignant melanomas in Xiphophorus hybrid tumor models. The CDKN2-related sequence we have identified and linked to melanoma induction is behaving as would be predicted for a tumor susceptibility gene; its identification with DIFF relies on very strong genetic linkage and map order data with LG V markers (Table 1, Fig. 3), and the compelling association of CDKN2L1 genotypes with susceptibility to UV-B-induced melanomas (Table 3). However, a two-gene inheritance model involving DIFF as a genetic trait determining pigment pattern phenotypes, as defined in the Sd, Gordon–Kosswig platyfish–swordtail hybrid, is not supported by our genetic linkage results for the Sp platyfish–swordtail hybrid. This finding suggests that proliferation and invasive tumor formation are separable in Xiphophorus melanoma models, and that other genes, in addition to DIFF, may regulate melanocyte proliferation; the identification of these genes will be an important focus of future studies.
In a recent study, Schartl et al. (53) investigated spontaneous melanoma formation in nonhybrid Xiphophorus species. In melanomas originating in certain purebred stocks, Xmrk overexpression was observed, consistent with a unifying mechanism for tumorigenesis in both hybrid and nonhybrid tumor models. However, the incidences of spontaneous melanoma formation in nonhybrid strains varies widely, from less than 1% to more than 25% (4, 23, 53), suggesting that simply overcoming suppression by DIFF is unlikely to be a common causal mechanism of melanoma formation. Our results, establishing linkage of a CDKN2-related sequence to the DIFF region in LG V, and to UV-B-induced melanoma formation in Sp platyfish–swordtail hybrids, support the role of DIFF as a critical genetic determinant of tumor formation. In future experiments, it will be important to determine if the CDKN2-related sequence we have cloned and mapped is also genotypically associated with spontaneous and induced tumor formation in other Xiphophorus melanoma models (especially the Gordon–Kosswig model). Determination of the complete structure of the LG V CDKN2 and its immediate genomic region in X. maculatus as well as other Xiphophorus species, and its gene expression characteristics in tissues and tumors from parental and hybrid animals, will lead to a more detailed and fundamental understanding of the genetic basis of these unique, heritable tumor models.
Acknowledgments
We especially thank Dr. Avril Woodhead (Brookhaven National Laboratory, Upton, NY) for performing histological evaluation of tumors. We are also grateful for the technical assistance of JoAnne Lund and Lela Limmer. This work was supported by Public Health Service Grant CA55245 from the National Cancer Institute and Grant ARP-029 from the Texas Higher Education Coordinating Board. S.K. is a postdoctoral fellow supported by National Cancer Institute Training Grant CA09480.
Footnotes
References
- 1.Anders F, Diehl H, Schwab M, Anders A. In: Pigment Cell. Klaus N, editor. Vol. 4. Basel: Karger; 1979. pp. 142–149. [Google Scholar]
- 2.Gordon M. Proc Natl Acad Sci USA. 1931;17:276–280. doi: 10.1073/pnas.17.5.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anders A, Anders F. Biochim Biophys Acta. 1978;516:61–95. doi: 10.1016/0304-419x(78)90004-5. [DOI] [PubMed] [Google Scholar]
- 4.Vielkind J R, Kallman K D, Morizot D C. J Aquat Anim Health. 1989;1:69–77. [Google Scholar]
- 5.Schartl M. Trends Genet. 1995;11:185–189. doi: 10.1016/S0168-9525(00)89041-1. [DOI] [PubMed] [Google Scholar]
- 6.Vielkind U. J Exp Zool. 1976;196:197–204. doi: 10.1002/jez.1401960207. [DOI] [PubMed] [Google Scholar]
- 7.Wittbrodt J, Adam D, Malitschek B, Maueler W, Raulf F, Telling A, Robertson S M, Schartl M. Nature (London) 1989;341:415–421. doi: 10.1038/341415a0. [DOI] [PubMed] [Google Scholar]
- 8.Adam D, Dimitrijevic N, Schartl M. Science. 1992;259:816–819. doi: 10.1126/science.8430335. [DOI] [PubMed] [Google Scholar]
- 9.Zechel C, Schleenbecker U, Anders A, Anders F. Oncogene. 1988;3:605–617. [PubMed] [Google Scholar]
- 10.Woolcock B W, Schmidt B M, Kallman K D, Vielkind J R. Cell Growth Differ. 1994;5:575–583. [PubMed] [Google Scholar]
- 11.Schartl M. Genetics. 1990;126:1083–1091. doi: 10.1093/genetics/126.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Winkler C, Wittbrodt J, Lammers R, Ullrich A, Schartl M. Oncogene. 1994;9:1517–1525. [PubMed] [Google Scholar]
- 13.Gruis N A, van der Velden P A, Sandkuijl L A, Prins D E, Weaver-Feldhaus J, Kamb A, Bergman W, Frants R R. Nat Genet. 1995;10:351–353. doi: 10.1038/ng0795-351. [DOI] [PubMed] [Google Scholar]
- 14.Gruis N A, Sandkuijl L A, van der Velden P A, Bergman W, Frants R R. Melanoma Res. 1995;5:169–177. doi: 10.1097/00008390-199506000-00005. [DOI] [PubMed] [Google Scholar]
- 15.Cairns P, Polascik T J, Eby Y, Tokino K, Califano J, et al. Nat Genet. 1995;11:210–212. doi: 10.1038/ng1095-210. [DOI] [PubMed] [Google Scholar]
- 16.Liu L, Lassam N J, Slingerland J M, Bailey D, Cole D, Jenkins R, Hogg D. Oncogene. 1995;11:405–412. [PubMed] [Google Scholar]
- 17.Reed J A, Loganzo F, Jr, Shea C R, Walker G J, Flores J F, Glendening J M, Bogdany J K, Shiel M J, Haluska F G, Fountain J W, Albino A P. Cancer Res. 1995;55:2713–2718. [PubMed] [Google Scholar]
- 18.Pollock P M, Yu F, Qiu L, Parsons P G, Hayward N K. Oncogene. 1995;11:663–668. [PubMed] [Google Scholar]
- 19.Setlow R B, Woodhead A D, Grist E. Proc Natl Acad Sci USA. 1989;86:8922–8926. doi: 10.1073/pnas.86.22.8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Setlow R B, Grist E, Thompson K, Woodhead A D. Proc Natl Acad Sci USA. 1993;90:6666–6670. doi: 10.1073/pnas.90.14.6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kallman K D. In: Handbook of Genetics. King R C, editor. Vol. 4. New York: Plenum; 1975. pp. 81–132. [Google Scholar]
- 22.Clark E. Science. 1950;112:722–723. doi: 10.1126/science.112.2920.722. [DOI] [PubMed] [Google Scholar]
- 23.Kazianis S, Borowsky R. J Hered. 1995;86:199–203. doi: 10.1093/oxfordjournals.jhered.a111562. [DOI] [PubMed] [Google Scholar]
- 24.Serrano M, Hannon G J, Beach D. Nature (London) 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 25.Okamoto A, Demetrick D J, Spillare E A, Hagiwara K, Hussain S P, Bennett W P, Forrester K, Gerwin B, Serrano M, Beach D H, Harris C C. Proc Natl Acad Sci USA. 1994;91:11045–11049. doi: 10.1073/pnas.91.23.11045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guan K L, Jenkins C W, Li Y, Nichols M A, Wu X, O’Keefe C L, Matera A G, Xiong Y. Genes Dev. 1994;8:2939–2952. doi: 10.1101/gad.8.24.2939. [DOI] [PubMed] [Google Scholar]
- 27.Don R H, Cox P T, Wainwright B J, Baker K, Mattick J S. Nucleic Acids Res. 1991;19:5081. [Google Scholar]
- 28.Roux K H. BioTechniques. 1994;16:812–814. [PubMed] [Google Scholar]
- 29.Sanger F, Nicklen S, Coulson A R. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morizot D C, Wright D A, Siciliano M J. Genetics. 1977;86:645–656. doi: 10.1093/genetics/86.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morizot D C, Schmidt M E. In: Applications of Electrophoresis and Isoelectric Focusing in Fisheries Management. Whitmore D H, editor. Boca Raton, FL: CRC; 1990. pp. 23–80. [Google Scholar]
- 32.Harless J, Svensson R, Kallman K D, Morizot D C, Nairn R S. Cancer Genet Cytogenet. 1990;50:45–51. doi: 10.1016/0165-4608(90)90236-4. [DOI] [PubMed] [Google Scholar]
- 33.Walter R B, Rolig R L, Kozak K, McEntire B, Morizot D C, Nairn R S. Mol Biol Evol. 1993;10:1227–1238. doi: 10.1093/oxfordjournals.molbev.a040066. [DOI] [PubMed] [Google Scholar]
- 34.Nairn R S, Della-Coletta L, McEntire B B, Walter R B, Morizot D C. Cancer Genet Cytogenet. 1996;88:144–150. doi: 10.1016/0165-4608(95)00296-0. [DOI] [PubMed] [Google Scholar]
- 35.Morizot D C, Harless J, Nairn R S, Kallman K D, Walter R B. In: Genetic Maps. 6th Ed. O’Brien S J, editor. Plainview, NY: Cold Spring Harbor Lab. Press; 1993. pp. 4.318–4.325. [Google Scholar]
- 36.McAlpine P J, Boucheix C, Pakstis A J, Strane L C, Berent T G, Shows T B. Cytogenet Cell Genet. 1988;49:4–45. doi: 10.1159/000132645. [DOI] [PubMed] [Google Scholar]
- 37.Morizot D C, Slaugenhaupt S A, Kallman K D, Chakravarti A. Genetics. 1991;127:399–410. doi: 10.1093/genetics/127.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lander E S, Green P, Abrahamson J, Barlow A, Daly M J, Lincoln S E, Newburg L. Genomics. 1987;1:174–181. doi: 10.1016/0888-7543(87)90010-3. [DOI] [PubMed] [Google Scholar]
- 39.Chan F K, Zhang J, Cheng L, Shapiro D N, Winoto A. Mol Cell Biol. 1995;15:2682–2688. doi: 10.1128/mcb.15.5.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lambert S, Yu H, Prchal J T, Lawler J, Ruff P, Speicher D, Cheung M C, Kan Y W, Palek J. Proc Natl Acad Sci USA. 1990;87:1730–1734. doi: 10.1073/pnas.87.5.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rigel D S, Kopf A W, Friedman R J. J Am Acad Dermatol. 1987;17:1050–1053. doi: 10.1016/s0190-9622(87)80487-5. [DOI] [PubMed] [Google Scholar]
- 42.Koh H K. N Engl J Med. 1991;325:171–182. doi: 10.1056/NEJM199107183250306. [DOI] [PubMed] [Google Scholar]
- 43.deGruijl F R, Van der Leun J C. In: Environmental UV Photobiology. Young A R, Bjorn L O, Moan J, Nultsch W, editors. New York: Plenum; 1993. pp. 89–112. [Google Scholar]
- 44.Meyer L J, Zone J H. J Invest Dermatol. 1994;103:112S–116S. doi: 10.1111/1523-1747.ep12399409. [DOI] [PubMed] [Google Scholar]
- 45.Koh H K, Kligler B E, Lew R A. Photochem Photobiol. 1990;51:765–779. [PubMed] [Google Scholar]
- 46.Kamb A, Shattuck-Evans D, Eeles R, Liu Q, Gruis N A, et al. Nat Genet. 1994;8:22–27. doi: 10.1038/ng0994-22. [DOI] [PubMed] [Google Scholar]
- 47.Thompson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morizot D C, Siciliano M J. J Natl Cancer Inst. 1983;71:809–813. [PubMed] [Google Scholar]
- 49.Ahuja M R, Schwab M, Anders F. J Hered. 1980;71:403–407. doi: 10.1093/oxfordjournals.jhered.a109395. [DOI] [PubMed] [Google Scholar]
- 50.Fornzler D, Wittbrodt J, Schartl M. Biochem Genet. 1991;29:11–12. doi: 10.1007/BF02426867. [DOI] [PubMed] [Google Scholar]
- 51.Kazianis S, Morizot D C, McEntire B B, Nairn R S, Borowsky R L. Genome Res. 1996;6:280–289. doi: 10.1101/gr.6.4.280. [DOI] [PubMed] [Google Scholar]
- 52.Nairn R S, Morizot D C, Kazianis S, Woodhead A D, Setlow R B. Photochem Photobiol. 1996;64:440–448. doi: 10.1111/j.1751-1097.1996.tb03089.x. [DOI] [PubMed] [Google Scholar]
- 53.Schartl A, Malitschek B, Kazianis S, Borowsky R, Schartl M. Cancer Res. 1995;55:159–165. [PubMed] [Google Scholar]