

Abstract
Biochemical studies have shown that the periplasmic protein disulfide oxidoreductase DsbC can isomerize aberrant disulfide bonds. Here we present the first evidence for an in vivo role of DsbC in disulfide bond isomerization. Furthermore, our data suggest that the enzymes DsbA and DsbC play distinct roles in the cell in disulfide bond formation and isomerization, respectively. We have shown that mutants in dsbC display a defect in disulfide bond formation specific for proteins with multiple disulfide bonds. The defect can be complemented by the addition of reduced dithiothreitol to the medium, suggesting that absence of DsbC results in accumulation of misoxidized proteins. Mutations in the dipZ and trxA genes have similar phenotypes. We propose that DipZ, a cytoplasmic membrane protein with a thioredoxin-like domain, and thioredoxin, the product of the trxA gene, are components of a pathway for maintaining DsbC active as a protein disulfide bond isomerase.
Keywords: DsbA, DsbC, protein folding
Disulfide bond formation has been studied for the most part in in vitro systems using model proteins such as RNase A, bovine pancreatic trypsin inhibitor (BPTI), and α-lactalbumin. These studies have raised two important questions concerning efficient disulfide bond formation. First, the rate of disulfide bond formation is slow in vitro, suggesting that, in vivo, catalysts may be required. Second, in proteins with multiple disulfide bonds, incorrect disulfide bonds can form, suggesting the need for an isomerase activity. The best characterized disulfide bond formation pathway is that of BPTI, a small, 58 amino acid polypeptide with 3 disulfide bonds in its native conformation (1). In the case of BPTI, folding intermediates occur in which non-native disulfide bonds form. Creighton and coworkers (1, 2) have proposed that these non-native disulfide bonds are an integral part of the disulfide folding pathway of BPTI. According to this proposal, the efficient folding of BPTI requires that disulfide linkages be broken and new ones formed, in effect “shuffling,” or isomerizing the disulfide bonds.
In Escherichia coli, disulfide bond formation occurs in the periplasm. The main enzyme catalyzing disulfide bond formation is DsbA, a 23-kDa protein with a thioredoxin-like fold (3, 4). Mutations in dsbA cause a pleiotropic defect in disulfide bond formation, resulting in loss of motility, reduced alkaline phosphatase activity, the inability to assemble F pili and Pap pili, as well as increased sensitivity to the reducing agent dithiothreitol (DTT) (3, 5). After catalyzing the formation of a disulfide bond, DsbA is left reduced and must be re-oxidized. The latter function is thought to be performed by DsbB, an integral membrane protein residing in the cytoplasmic membrane (3, 5–7, 34).
Recently, a second periplasmic disulfide bond oxidoreductase, DsbC, was discovered in both E. coli and Erwinia chrysanthemi (8, 9). The data concerning the in vivo phenotypes of a dsbC null are rather confusing. Although no great defect in disulfide bond formation was detectable in Erwinia, the E. coli dsbC null was reported to have a decrease in disulfide bond formation similar to that observed in a dsbA mutant. In vitro studies have shown that DsbC has the ability to isomerize disulfide bonds in folding intermediates of BPTI, a reaction that DsbA is very poor at catalyzing (10). These findings suggested that DsbC may be required for disulfide bond isomerization.
Here we report results that support a role for DsbC as a disulfide bond isomerase in vivo. Furthermore, our studies suggest that both the membrane protein DipZ, thought to be involved in cytochrome c biosynthesis (11, 12), and the cytoplasmic protein thioredoxin are required for the maintenance of DsbC as an isomerase. These findings arose from a study of suppressor mutations that increase the rate of disulfide bond formation in a dsbA mutant.
MATERIALS AND METHODS
Strains and Media.
The strains used in this study are listed in Table 1. Bacteria were grown in NZ medium (10 g Nzamine/8 g NaCl/5 g Yeast extract per liter), except for cultures grown for alkaline phosphatase assays and pulse-chase experiments, in which case the cells were grown in M63 minimal medium [13.6 g KH2PO4/2 g (NH4)2SO4/0.5 g FeSO4·7H2O/0.2 g MgSO4·7 H2O per liter, pH 7.0] with glucose as carbon source and supplemented with 18 amino acids (-cysteine, -methionine).
Table 1.
Strains used in this study
| Strain | Genotype | Source or ref. |
|---|---|---|
| MC1000 | araD139 Δ(araABC-leu)7679 galU galK Δ(lac)X74 rpsL thi | Lab collection |
| E2088 | F-araD139 Δ(ara-leu)797 Δ(proAB-argF-lacIPOZYA)XIII rpsL Nalr exa-1::MudI1734 cadC1:Tn10 | (Ref. 13) |
| SM594 | MC1000 phoR phoA68 | (Ref. 14) |
| RI27 | SM594 dsbA::kan1 | This study |
| RI89 | MC1000 phoR Δara714 leu+ | This study |
| RI90 | RI89 dsbA::kan1 | This study |
| RI92 | MC1000 phoR Δara714 leu+phoA68 | This study |
| RI93 | RI92 dsbA::kan1 | This study |
| RI179 | RI89 ΔdsbC::cam | This study |
| RI181 | RI89 ΔdsbC::cam dsbA::kan1 | This study |
| A307 | ΔtrxA307 | (Ref. 15) |
| RI258 | RI89 ΔtrxA307 | This study |
| RI266 | RI89 ΔtrxA307 ΔdsbC::cam | This study |
| RI242 | RI89 dipZ::mini-Tn10Cam1 | This study |
| RI247 | RI89 ΔdsbC::cam dipZ::mini-Tn10Cam1 cadC1::Tn10 | This study |
| SE600 | ΔompA | (Ref. 16) |
| RI216 | RI93 ΔompA zch:Tn10 | This study |
| RI288 | RI93 dipZ::mini-Tn10Cam1 ΔompA zch:Tn10 | This study |
| RI289 | RI93 dipZ::mini-Tn10Cam1 | This study |
| JCB495 | MC1000 recD | Lab collection |
For gene symbols, see ref. 17. kan, Kanamycin resistance; cam, chloramphenicol resistance.
Strain Construction and Plasmids.
Standard molecular and genetic techniques were used for strain and plasmid construction (18). Plasmid pJC763 (19) was used to construct the dsbC deletion. The plasmid was digested using KpnI and MluI releasing a 1.2-kb fragment containing the dsbC gene. The vector containing the flanking regions was isolated and the ends made blunt using the Klenow fragment of E. coli DNA polymerase I. The chloramphenicol resistance gene from the mini-Tn10 derivative of λNK1324 (20) was excised as a BamHI fragment, the ends of which were made blunt, as above, and cloned into the vector containing the DNA flanking dsbC, generating plasmid pJC763Δcam. The proper construction of the plasmid was checked by restriction digestion. The deletion of dsbC generated in this way was crossed onto the E. coli chromosome by linear transformation into the recD strain JCB495, which was performed as described (21). The correct replacement of dsbC in the deletion construct was confirmed using primers flanking the deleted DNA sequences. The dipZ gene was cloned into pBAD30 (22) on a 9.5-kb KpnI fragment from Kohara phage 648 (23), generating plasmid pDIPZ1. Plasmid pDIPZ2 was constructed by subcloning the 2.7-kb SalI/XmaI fragment containing the dipZ gene into pBAD30. The plasmid p30ompA was constructed by cloning the BclI/PstI fragment containing the ompA gene from plasmid pOMPAΔ115 (16) into the vector pWSK30 (24). The ompA gene was then cloned into pBAD22 as an XbaI/HindIII fragment, yielding plasmid p22ompA in which ompA expression is under control of the arabinose operon pBAD promoter. The cysteine mutation (cys310 → gly) was cloned into p22ompA by replacing the 640-bp SphI/HindIII fragment, representing the 3′ end of the ompA gene, with the SphI/HindIII fragment from pKT001 (25), carrying the mutation. The correct construction of the cysteine mutant plasmid was confirmed by restriction digestion; the cys → gly mutation introduced an HaeIII site. Expression of wild-type ompA and the cysteine mutant was confirmed by Western blot analysis and found to be equal to the expression of the chromosomal copy of ompA under fully inducing conditions (0.2% arabinose in NZ medium).
Transposon mutagenesis was performed as described (20). Transposon insertion mutants of RI216 were screened for increased motility and alkaline phosphatase activity. Insertion of the transposon in dipZ was confirmed by PCR. The exact sites of insertion in dipZ were determined by sequencing of the PCR product.
Enzymatic Assays.
Alkaline phosphatase assays were performed as described (26), except that cells were treated with 100 mM iodoacetamide on ice for 10 min prior to washing. The values shown represent the average of three separate cultures.
Urokinase assays were performed on strains transformed with plasmid pRDB8-A, which expresses mouse urokinase plasminogen activator constitutively (27). Whole cell lysates of bacteria grown to exponential phase were separated on non-reducing SDS/PAGE gels and urokinase activity was shown by assaying plasminogen activation using plasminogen/casein agar as described (28).
Pulse-Chase Analysis and Immunoprecipitation.
Bacteria were grown to early logarithmic phase at 37°C in M63 minimal medium with glucose as carbon source and supplemented with 18 amino acids. Cells were pulse-labeled for 30 sec with radioactive [35S]methionine, and chased by the addition of cold methionine to a final concentration of 0.1%. Samples were removed at various time points, placed on ice, and incubated in the presence of excess methionine and iodoacetamide (100 mM final concentration) for at least 15 min. The cells were then pelleted, resuspended in 40 μl of SDS buffer (1% SDS/10 mM Tris·Cl, pH 8.0/1 mM EDTA) and lysed by boiling for 3 min. The lysates were then diluted in KI buffer (2% Triton X-100/50 mM Tris·Cl, pH 8.0/150 mM NaCl/1 mM EDTA) and insoluble cellular debris was removed by centrifugation. The proteins of interest were immunoprecipitated as described (29).
RESULTS
Isolation of Mutations Suppressing Disulfide Bond Formation Defects in a dsbA Mutant.
We have sought mutants that restore disulfide bond formation in a dsbA null strain by screening for spontaneous mutants that had regained motility. To avoid mutations, which restore motility without restoring disulfide bond formation, we screened the motile mutants for increased alkaline phosphatase activity, another indicator of disulfide bond formation. We used a strain with a signal sequence mutation in alkaline phosphatase, phoA68, that reduces the amount of secreted alkaline phosphatase to about 16% of wild type (14), allowing easy distinction of dsbA− and wild-type bacteria on XP plates.
Of 89 motile revertants, 11 had increased alkaline phosphatase activity. The characteristics of the various suppressor mutant classes are listed in Table 2. We mapped the different suppressor loci using standard genetic techniques. The first class of mutations mapped just upstream of the cadC gene. The known sequence of this region contains a 1.4-kb open reading frame coding for DipZ, a cytoplasmic membrane protein involved in cytochrome c biogenesis with a thioredoxin-like domain, which is thought to be periplasmic. We cloned a 2.7-kb XmaI/SalI fragment, which carries the dipZ gene and found that it complemented the suppressor mutation (see Materials and Methods). To test whether a null mutation of dipZ can suppress the dsbA− phenotype, we isolated transposon insertions in dipZ. The sites of insertion were determined by sequencing out of the transposon and determined to be in codons 3, 108, 350, and 463 of the dipZ open reading frame. These transposon insertions suppressed the dsbA− phenotype in a manner similar to the original point mutation (data not shown). Missiakas et al. (30) have reported that strains mutant in dipZ(dsbD) suppress a dsbA null with respect to DTT sensitivity. The dipZ gene is also involved in copper tolerance (31). Our dipZ mutants are sensitive to copper; the parental dsbA mutant strain grew in the presence of 4 mM CuSO4, whereas the dsbA/dipZ double mutant did not (Table 2).
Table 2.
Characteristics of the three mutant classes
| Strain | Alkaline phosphatase activity* | ompA† | Motility | Cu2+ resistance‡ | DsbC dependence§ | Locus | No. of mutants |
|---|---|---|---|---|---|---|---|
| Wild type | 159.3 ± 12.4 | + | ++++ | + | |||
| dsbA::kan | 4 ± 1.8 | + | — | + | |||
| Class I | 7.9 ± 1.6 | + | + | — | Yes | dipZ− | 5 |
| Class II | 22.2 ± 4.9 | — | +++ | — | Yes | dipZ− | 2 |
| Class III | 10.5 ± 4.9 | + | + | −/+ | Yes | trxA− | 1 |
| Class II/dsbC− | 2.2 ± 1.6 | — | |||||
| Class III/dsbC− | Not assayed | — |
Phosphatase assays were performed as described in Materials and Methods on strains carrying the phoA68 signal sequence mutation.
Indicates whether or not the strains contain an intact ompA gene.
Cu2+ resistance refers to the ability of the strain to grow in the presence of 4 mM cupric sulfate.
DsbC dependence indicates whether suppression depends on the presence of a functional dsbC allele.
A second suppressor mutation that did not map to dipZ was also slightly copper sensitive. We cloned the wild-type gene for this mutation by selecting for a plasmid that allowed growth in the presence of copper using an HindIII library of the parental dsbA mutant cloned in pBAD24. The complementing clones contained a chromosomal fragment spanning the trxA gene, coding for thioredoxin. The mutant strain was resistant to killing by phage T7, which requires thioredoxin for efficient replication. A plasmid containing only the trxA gene cloned under control of the arabinose promoter complemented the mutant for both the suppression of motility and copper sensitivity, confirming that a mutation in trxA was responsible for the suppressor phenotype. In addition, a deletion spanning the trxA gene (15) suppressed the dsbA null phenotype.
While analyzing the effect of the suppressor mutations on the rate of disulfide bond formation, we noticed that two of the seven dipZ mutants did not express any immunoprecipitable OmpA. OmpA is a major outer membrane protein with a single disulfide bond in its periplasmic C-terminal domain. The strains lacking OmpA displayed increased motility and alkaline phosphatase activity compared with the suppressor mutants that were OmpA+ (Table 2). Crossing back the wild-type dipZ allele into these mutants abolished suppression, whereas introducing a wild-type copy of ompA changed the phenotype to that of the simple dipZ suppressor. Deleting ompA by itself did not suppress the dsbA mutant phenotype significantly (no increase in alkaline phosphatase activity and only a very slight restoration of motility were detectable), indicating that it was only able to enhance the effectivity of a second suppressor mutation. We reasoned that OmpA, due to its high level of expression, is a major substrate of the disulfide bond-forming machinery and is, therefore, a drain on the remaining capacity to form disulfide bonds in a dsbA− strain. Indeed, although expression of wild-type ompA was able to change the suppression phenotype of the double mutant to that of the dipZ- ompA+ suppressor alone with respect to motility, expression of a mutant eliminating one of the cysteines of ompA had no effect (data not shown). This result is consistent with the proposal that the synergistic effect of deleting ompA on suppression by the dipZ null is due to the removal of a highly expressed substrate for disulfide bond formation.
Suppression Depends on the Presence of DsbC.
Overexpression of DsbC can complement the defect in disulfide bond formation in a dsbA mutant (8, 9). This finding suggested to us that the suppression we are observing might work via DsbC. We found that introduction of a dsbC null mutation in which the gene was deleted and replaced by a chloramphenicol cassette into suppressor strains (see Materials and Methods) abolished the restoration of both motility and alkaline phosphatase activity (Table 2). Because suppression was DsbC dependent, it seemed possible that DsbC was also responsible for the residual disulfide bond formation observed in a dsbA mutant strain. However, deletion of dsbC had no effect on motility (data not shown), nor did it greatly affect alkaline phosphatase activity of either the wild-type or the dsbA null strain (Table 3, phoA+ strains were assayed). To further analyze disulfide bond formation in these strains we performed pulse-chase experiments (Fig. 1). In the case of the wild type, disulfide bond formation is completed at the 1-min time point (Fig. 1). Comparison of the dsbA mutant and the dsbA/dsbC double-mutant strains revealed little if any difference in the rate of disulfide bond formation of OmpA or alkaline phosphatase, which was rapidly degraded (Fig. 1).
Table 3.
Effect of deleting dsbC on the alkaline phosphatase activity in wild-type and dsbA− E. coli
| Strain | Alkaline phosphatase activity* |
|---|---|
| Wild type | 811 ± 42 |
| ΔdsbC::cam | 777 ± 53 |
| dsbA::kan | 14.4 ± 2 |
| dsbA::kan/ΔdsbC::cam | 20.3 ± 4.2 |
Phosphatase activity was measured as described in the Materials and Methods.
Figure 1.
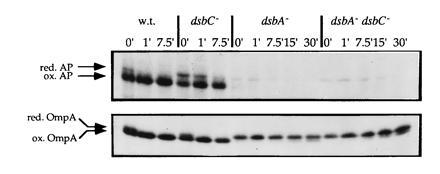
Disulfide bond formation in alkaline phosphatase and OmpA in wild-type, dsbC−, dsbA−, and dsbA−/dsbC− strains. Pulse-chase experiments were performed as described. The positions of reduced and oxidized alkaline phosphatase and OmpA are indicated. The strains used were RI89 (wild type), RI179 (ΔdsbC::cam), RI90 (dsbA::kan1), and RI181 (ΔdsbC::cam dsbA::kan1).
A dsbC Mutant Does Affect Alkaline Phosphatase Maturation.
The pulse-chase experiment revealed that the dsbC null by itself does have an effect on disulfide bond formation in alkaline phosphatase (Fig. 1 and see below). The defect is not great (quantitation showed that only about 15% of the protein is not properly oxidized at the 1-min time point) and does not greatly affect the overall alkaline phosphatase activity of the strain (Table 3). In contrast, the formation of the disulfide bond in OmpA is not affected in the dsbC mutant strain. This would make sense if the function of DsbC in vivo is indeed that of a disulfide bond isomerase as the biochemical studies suggest, since alkaline phosphatase has two disulfide bonds and could potentially be misoxidized, whereas OmpA only has a single disulfide bond.
As an isomerase, one would expect that DsbC must be maintained in a reduced state to attack disulfide bonds in misoxidized proteins. One could postulate that the reason for increased oxidation in the dipZ null is due to a rise in the level of oxidized DsbC in this strain, which can in turn catalyze de novo disulfide bond formation. If this is the case, then DipZ could play a role in an isomerization pathway by keeping DsbC reduced in the otherwise oxidizing periplasm.
dipZ and trxA Mutants Also Have Defects in Alkaline Phosphatase Folding.
If DipZ is required for DsbC to function properly, one would expect a dipZ mutant to display a defect in alkaline phosphatase folding similar to that of a dsbC mutant. This is the case (Fig. 2). Moreover, the effects of the mutations are not cumulative (Fig. 2), consistent with the proposal that DipZ and DsbC act in a pathway. Pulse-chase experiments showed that the thioredoxin mutation affects alkaline phosphatase folding, albeit to a lesser extent than the dsbC mutation (data not shown).
Figure 2.
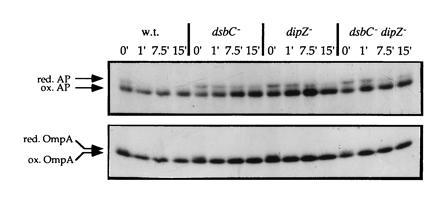
Effect of the dipZ::miniTn10Cam insertion on folding of alkaline phosphatase and OmpA. Samples were treated as described. The positions of reduced and oxidized alkaline phosphatase and OmpA are indicated. The strains used were RI89 (wild type), RI179 (ΔdsbC::cam), RI242 (dipZ::mini-Tn10Cam1), and RI247 (ΔdsbC::cam dipZ::mini-Tn10Cam1).
The Defect in Alkaline Phosphatase Folding Can Be Complemented by the Addition of Reduced DTT to Growing Cells.
The defect in cytochrome c biogenesis in a dipZ mutant can be complemented by certain reducing agents (16), and our proposed role for DipZ in disulfide bond isomerization is also a reductive one. Therefore, we asked whether reducing agents can complement a trxA, a dipZ, or a dsbC mutant. Addition of 50 μM reduced DTT to the medium 5–10 min before labeling was able to complement the defect in alkaline phosphatase folding in the trxA−, dipZ−, and dsbC− strains (Fig. 3). Oxidized DTT cannot complement the folding defect (data not shown and see below results with urokinase).
Figure 3.
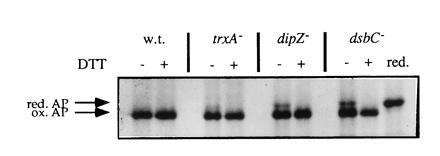
Complementation of the trxA−, dipZ−, and dsbC− mutations by addition of reduced DTT to the medium. Strains were grown to early logarithmic phase, labeled with [35S]methionine for 30 sec, followed by a 1-min chase with unlabeled methionine at which point samples were removed and placed on ice in the presence of 100 mM iodoacetamide. Immunoprecipitation of alkaline phosphatase and gel electrophoresis were performed as described. Reduced DTT was added to the medium 5–10 min prior to labeling in the lanes designated with a +. In the lane designated red., the sample of the dsbC mutant that had not been treated with DTT during growth was reduced with 10 mM DTT at 80°C and alkylated with an excess of iodoacetamide at room temperature prior to loading on the gel.
We showed that the band we have suggested to be misoxidized alkaline phosphatase was not the precursor of alkaline phosphatase carrying its signal sequence by reducing the sample from the dsbC null-strain grown in the absence of DTT. Reduction resulted in only a single band, corresponding to mature alkaline phosphatase (Fig. 3). These results support the idea that the slower migrating species of alkaline phosphatase is indeed not simply a reduced species of alkaline phosphatase, but a misoxidized variant. We suggest that DTT added to growing cells is reducing the aberrant disulfide bond or bonds, giving the protein another opportunity to fold properly.
Urokinase Does Not Fold Properly in dsbC, dipZ, and trxA Mutants.
Mouse urokinase has 12 disulfide bonds, 6 of which are in the serine protease domain. One would expect that the chance that non-native disulfide bonds are formed increases with the number of disulfide bonds and that the requirement for a functional isomerase would be greater for proteins with multiple disulfide bonds. Urokinase can be expressed and secreted in E. coli and its activity observed in a zymogram, assaying its ability to activate plasminogen (28). Both the dipZ and the dsbC mutants displayed a greater than 100-fold reduction in urokinase activity (Fig. 4A). The trxA mutation causes urokinase activity to decrease to about 5% of wild type (as determined by comparison with dilutions of the wild-type extract). These data are consistent with the results of the alkaline phosphatase pulse-chase experiments, where the effect of the trxA deletion was less pronounced than that of the dipZ and dsbC mutations. Thus, while the dsbC mutation has no great effect on alkaline phosphatase activity, urokinase with its greater number of disulfide bonds is severely affected.
Figure 4.
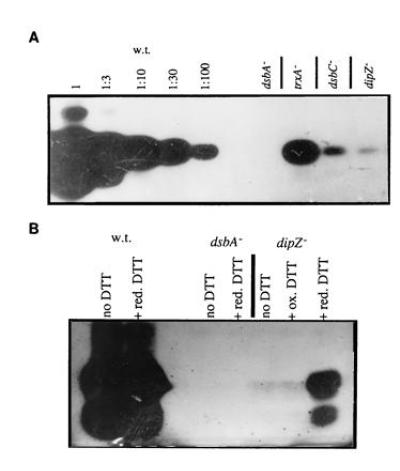
Effect of dsbA, ΔtrxA, dipZ, and dsbC mutations on urokinase activity. Lysates of strains grown to mid-logarithmic phase were separated on non-reducing SDS/PAGE gels and urokinase activity was shown by activation of plasminogen on plasminogen/casein agar. Zymograms of lysates from strains RI89 (wild type), RI90 (dsbA::kan1), RI258 (ΔtrxA), RI242 (dipZ::mini-Tn10Cam1), and RI179 (ΔdsbC::cam) are shown in A. Relative activities of the mutants were estimated by comparison to dilutions of the wild-type lysate. (B) The indicated strains were grown without DTT, in the presence of 1 mM reduced DTT (red. DTT) or 1 mM oxidized DTT (ox. DTT).
We also examined whether reduced DTT can complement the defect in urokinase folding when added to growing cells (Fig. 4B). A higher concentration of reduced DTT (1 mM) was used in this experiment because the concentration added to the cells in the pulse-chase experiments was not sufficient to suppress the urokinase defect. This is presumably due to air oxidation of the reductant, which was added at the beginning of the incubation, instead of just before sample processing, as was the case when analyzing alkaline phosphatase. Reduced DTT had no effect on urokinase activity in the wild-type strain, nor did it restore urokinase activity in the dsbA null. However, reduced, but not oxidized, DTT partially restored urokinase activity in the dipZ null. The urokinase defect of the dsbC mutant could be complemented by reduced DTT as well. The results were quite similar to those seen with the dipZ strain (data not shown).
DISCUSSION
We have presented the first in vivo evidence for a role of DsbC as a disulfide bond isomerase. Furthermore, we have presented results that suggest that thioredoxin, DipZ, and DsbC are components of a pathway for disulfide bond isomerization in vivo. Mutations in the corresponding genes affect disulfide bond formation in proteins with multiple disulfide bonds. Moreover, the severity of the defect appears to increase with the number of disulfide bonds. Our data suggest that the misfolded alkaline phosphatase detected in dipZ, dsbC, and trxA mutants, while having the same relative mobility as reduced alkaline phosphatase, is actually a misoxidized species, whose accumulation can be prevented by the addition of low concentrations of reduced DTT to growing cells. It is difficult to see how the reductant, DTT, could enhance disulfide bond formation in a reduced version of alkaline phosphatase, whereas DTT could substitute for DsbC by reducing incorrect disulfide bonds and giving DsbA another opportunity to catalyze formation of the correct cysteine linkages. Although these results raise the possibility that DsbC is simply part of a periplasmic reduction system, the in vitro data that shows that DsbC is able to catalyze disulfide bond isomerization suggest a direct role for DsbC as an isomerase.
The proposed function of DsbC as an isomerase would require that the active site of DsbC be maintained in a reduced state to be able to attack non-native disulfide bonds and isomerize them. Although the oxidation state of DsbC should not be altered by the isomerization process, the requirement that DsbC be reduced poses a dilemma, since the periplasm of E. coli is a highly oxidizing environment. The results of the suppressor analysis, as well as the effects of the single mutations indicate that thioredoxin and DipZ may be components of a pathway involved in reducing DsbC. We favor the idea that DipZ is simply required to maintain DsbC in a reduced, active state as an isomerase, although we cannot rule out the possibility that DipZ actively participates in the process of disulfide bond isomerization.
The involvement of thioredoxin in this process is intriguing. We find that thioredoxin does not affect either dipZ or dsbC expression (unpublished results). One interesting possibility is that it contributes its reducing potential directly to the pathway leading to DsbC activity. It is not clear whether any of these three components interact directly. Thioredoxin may act directly or through an intermediate to reduce DipZ. In any case, how the reducing potential is transported from the cytoplasm across the membrane into the periplasm is an interesting subject for further study.
DsbC does not, from our studies, appear to be involved in de novo disulfide bond formation in a wild-type strain. Introduction of a deletion of the dsbC gene did not affect disulfide bond formation in OmpA, nor did it decrease motility. Furthermore, the defect in disulfide bond formation in a dsbA mutant strain was not enhanced by combining the dsbA null mutation with the dsbC deletion. These data contradict the earlier suggestion that strains mutant in dsbC display a defect in disulfide bond formation similar to dsbA mutants (8). We find it hard to reconcile these discrepancies although they may be due to strain differences or differences in experimental conditions. However, our findings are consistent with data from studies in Erwinia, which also identified DsbA as the main enzyme responsible for disulfide bond formation (9, 33). Our data, therefore, support the notion that DsbA is the main oxidase in the periplasm, whereas DsbC is required for the isomerization of disulfide bonds (Fig. 5).
Figure 5.
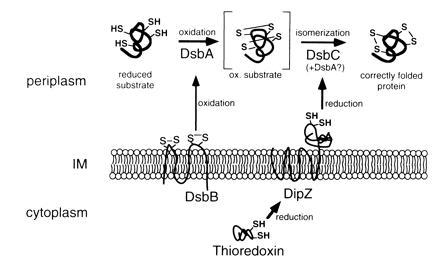
Model of disulfide bond formation in the E. coli periplasm. DsbA may be involved in disulfide bond isomerization, because there is a possibility that DsbC acts to reduce misoxidized proteins allowing DsbA to form the correct disulfide. The topology of DipZ presented in the model is based on the published model derived from computer predictions (11).
Our initial interest in looking for suppressors that restore disulfide bond formation in a dsbA background was rooted in the observation that disrupting the dsbA gene did not completely abolish disulfide bond formation. Instead of obtaining mutations that increased expression or activity of a second catalytic system, the suppressor mutations abolished expression of proteins thought to have a role as reducing agents in vivo. We suggest that eliminating DipZ or thioredoxin, which may participate in a pathway for reducing DsbC, causes a larger portion of the DsbC molecules to be in an oxidized state, where they in turn can act to catalyze the formation of disulfide bonds.
Our studies have not elucidated the nature of the residual disulfide bond forming activity in the dsbA− background. One possibility is that the residual activity is due to air oxidation. One could, however, also argue that we simply did not obtain mutations increasing either the activity or expression of a second system because this type of mutation is rarer than the null mutations we obtained in our screen.
The finding that deleting ompA synergistically enhances the efficiency of the suppressor mutations suggests that the potential to form disulfide bonds in the periplasm of a dsbA mutant is already extremely limited. If this were not the case, one would expect that simply deleting ompA should be able to suppress a dsbA null, since removing a major substrate of disulfide bond formation should relieve the strain on the remaining capacity to form disulfide bonds regardless of whether the strain carries a suppressor mutation or not.
In summary, based on the data presented in this paper, we conclude that DsbA is the main oxidant in the periplasm responsible for disulfide bond formation. In addition, we propose a pathway for disulfide bond isomerization, in which DsbC is the active isomerase and DipZ is required to keep DsbC in an active, reduced state. This study also suggests that the reducing power for DipZ may come from the cytoplasm via thioredoxin.
Acknowledgments
We thank Shoji Mizushima for providing plasmid pKT001, Joel Belasco for plasmid pOMPAΔ115 and strain SE600, Susan Lovett for plasmid pJC763, and Eric Olson for strain E2088. We gratefully acknowledge members of the Beckwith laboratory, in particular Will Prinz and Christophe Guilhot, for helpful discussions. We also thank Terri Luna for media preparation and Ann McIntosh for administrative assistance. This work was supported by grants from the American Cancer Society and National Science Foundation to J.B. and a grant from the Swiss National Science Foundation to D.B. J.B. is an American Cancer Society Research Professor.
Footnotes
Abbreviation: BPTI, bovine pancreatic trypsin inhibitor.
References
- 1.Creighton T E. Science. 1992;256:111–114. doi: 10.1126/science.1373519. [DOI] [PubMed] [Google Scholar]
- 2.Darby N J, Morin P E, Talbo G, Creighton T E. J Mol Biol. 1995;249:463–477. doi: 10.1006/jmbi.1995.0309. [DOI] [PubMed] [Google Scholar]
- 3.Bardwell J C, McGovern K, Beckwith J. Cell. 1991;67:581–589. doi: 10.1016/0092-8674(91)90532-4. [DOI] [PubMed] [Google Scholar]
- 4.Kamitani S, Akiyama Y, Ito K. EMBO J. 1992;11:57–62. doi: 10.1002/j.1460-2075.1992.tb05027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Missiakas D, Georgopoulos C, Raina S. Proc Natl Acad Sci USA. 1993;90:7084–7088. doi: 10.1073/pnas.90.15.7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guilhot C, Jander G, Martin N L, Beckwith J. Proc Natl Acad Sci USA. 1995;92:9895–9899. doi: 10.1073/pnas.92.21.9895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kishigami S, Kanaya E, Kikuchi M, Ito K. J Biol Chem. 1995;270:17072–17074. doi: 10.1074/jbc.270.29.17072. [DOI] [PubMed] [Google Scholar]
- 8.Missiakas D, Georgopoulos C, Raina S. EMBO J. 1994;13:2013–2020. doi: 10.1002/j.1460-2075.1994.tb06471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shevchik V E, Condemine G, Robert-Baudouy J. EMBO J. 1994;13:2007–2012. doi: 10.1002/j.1460-2075.1994.tb06470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zapun A, Missiakas D, Raina S, Creighton T E. Biochemistry. 1995;34:5075–5089. doi: 10.1021/bi00015a019. [DOI] [PubMed] [Google Scholar]
- 11.Crooke H, Cole J. Mol Microbiol. 1995;15:1139–1150. doi: 10.1111/j.1365-2958.1995.tb02287.x. [DOI] [PubMed] [Google Scholar]
- 12.Sambongi Y, Crooke H, Cole J A, Ferguson S J. FEBS Lett. 1994;344:207–210. doi: 10.1016/0014-5793(94)00399-8. [DOI] [PubMed] [Google Scholar]
- 13.Watson N, Dunyak D S, Rosey E L, Slonczewski J L, Olson E R. J Bacteriol. 1992;174:530–540. doi: 10.1128/jb.174.2.530-540.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michaelis S, Inouye H, Oliver D, Beckwith J. J Bacteriol. 1983;154:366–374. doi: 10.1128/jb.154.1.366-374.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russel M, Model P. J Biol Chem. 1986;261:14997–15005. [PubMed] [Google Scholar]
- 16.Emory S A, Belasco J G. J Bacteriol. 1990;172:4472–4481. doi: 10.1128/jb.172.8.4472-4481.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bachman B J. Microbiol Rev. 1990;54:130–197. doi: 10.1128/mr.54.2.130-197.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 19.Lovett S T, Kolodner R D. J Bacteriol. 1991;173:353–364. doi: 10.1128/jb.173.1.353-364.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleckner N, Bender J, Gottesman S. Methods Enzymol. 1991;204:139–180. doi: 10.1016/0076-6879(91)04009-d. [DOI] [PubMed] [Google Scholar]
- 21.Russel C B, Thaler D S, Dahlquist F W. J Bacteriol. 1989;171:2609–2613. doi: 10.1128/jb.171.5.2609-2613.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guzman L M, Belin D, Carson M J, Beckwith J. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohara Y, Akiyama K, Isono K. Cell. 1987;50:495–508. doi: 10.1016/0092-8674(87)90503-4. [DOI] [PubMed] [Google Scholar]
- 24.Wang R F, Kushner S R. Gene. 1991;100:195–199. [PubMed] [Google Scholar]
- 25.Tani K, Tokuda H, Mizushima S. J Biol Chem. 1990;265:17341–17347. [PubMed] [Google Scholar]
- 26.Brickman E, Beckwith J. J Mol Biol. 1975;96:307–316. doi: 10.1016/0022-2836(75)90350-2. [DOI] [PubMed] [Google Scholar]
- 27.Duvoisin R M, Belin D, Kirsch H M. Gene. 1986;45:193–201. doi: 10.1016/0378-1119(86)90254-4. [DOI] [PubMed] [Google Scholar]
- 28.Belin D, Vassalli J-D, Combepine C, Godeau F, Nagamine Y, Kocher E, Duvoisin R M. Eur J Biochem. 1985;148:225–232. doi: 10.1111/j.1432-1033.1985.tb08829.x. [DOI] [PubMed] [Google Scholar]
- 29.Froshauer S, Green G N, Boyd D, McGovern K, Beckwith J. J Mol Biol. 1988;200:501–511. doi: 10.1016/0022-2836(88)90539-6. [DOI] [PubMed] [Google Scholar]
- 30.Missiakas D, Schwager F, Raina S. EMBO J. 1995;14:3415–3424. doi: 10.1002/j.1460-2075.1995.tb07347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fong S T, Camakaris J, Lee B T. Mol Microbiol. 1995;15:1127–1137. doi: 10.1111/j.1365-2958.1995.tb02286.x. [DOI] [PubMed] [Google Scholar]
- 32.Sambongi Y, Ferguson S J. FEBS Lett. 1994;353:235–238. doi: 10.1016/0014-5793(94)01053-6. [DOI] [PubMed] [Google Scholar]
- 33.Shevchik V E, Bortoli-German I, Robert-Baudouy J, Robinet S, Barras F, Condemine G. Mol Microbiol. 1995;16:745–753. doi: 10.1111/j.1365-2958.1995.tb02435.x. [DOI] [PubMed] [Google Scholar]
- 34.Bardwell J C, Lee J O, Jander G, Martin N, Belin D, Beckwith J. Proc Natl Acad Sci USA. 1993;90:1038–1042. doi: 10.1073/pnas.90.3.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]