

Abstract
Bipolar mood disorder (BP) is a debilitating syndrome characterized by episodes of mania and depression. We designed a multistage study to detect all major loci predisposing to severe BP (termed BP-I) in two pedigrees drawn from the Central Valley of Costa Rica, where the population is largely descended from a few founders in the 16th–18th centuries. We considered only individuals with BP-I as affected and screened the genome for linkage with 473 microsatellite markers. We used a model for linkage analysis that incorporated a high phenocopy rate and a conservative estimate of penetrance. Our goal in this study was not to establish definitive linkage but rather to detect all regions possibly harboring major genes for BP-I in these pedigrees. To facilitate this aim, we evaluated the degree to which markers that were informative in our data set provided coverage of each genome region; we estimate that at least 94% of the genome has been covered, at a predesignated threshold determined through prior linkage simulation analyses. We report here the results of our genome screen for BP-I loci and indicate several regions that merit further study, including segments in 18q, 18p, and 11p, in which suggestive lod scores were observed for two or more contiguous markers. Isolated lod scores that exceeded our thresholds in one or both families also occurred on chromosomes 1, 2, 3, 4, 5, 7, 13, 15, 16, and 17. Interesting regions highlighted in this genome screen will be followed up using linkage disequilibrium (LD) methods.
Bipolar mood disorder (BP) is a debilitating syndrome characterized by episodes of mania and depression (1). Despite abundant evidence that BP has a major genetic component (2–5), linkage studies have not yet succeeded in definitively localizing a BP gene (6). This is chiefly because mapping studies of psychiatric disorders have generally been conducted under a paradigm that was appropriate for mapping genes for simple Mendelian disorders, namely, using linkage analysis in the expectation of finding high lod (logarithm of odds) scores that definitively signpost the location of disease genes. The follow-up to early BP linkage studies, however, showed that even extremely high lod scores at a single location can be false positives (7–10). The instability of the findings in these studies was at least partially due to the genetic models chosen for initial analyses, which incorporated broadly defined affected phenotypes, low phenocopy rates, and high penetrances; this approach set the stage for a precipitous diminution in the evidence for linkage when diagnoses for a few members of the original pedigrees changed over time (2, 5, 9, 10). In fact, the difficulty in defining behavioral phenotypes, their probable etiologic heterogeneity, and our lack of knowledge regarding their mode of transmission suggest that it is unlikely that any single linkage study will yield sufficient evidence to unequivocally localize a gene for a psychiatric disorder. Given this uncertainty, we propose that initial linkage studies be conducted using a conservative approach (affected phenotypes are narrowly defined and most linkage information derives from affected individuals) and that the results of lod score analysis be used for directing follow-up studies rather than as formal linkage tests. Despite the uncertainty in estimating genetic model parameters for complex traits, lod score analysis using large pedigrees is still an efficient means of initially screening the genome to detect hints of linkage over broad regions; as affected individuals in such pedigrees are separated from each other by only a few generations, few recombinations between a linked marker and a disease gene are likely to be observed over intervals of 10–15 centimorgans (cM) (the typical distances between markers used in our study) (11).
Fortunately, the recent availability of highly polymorphic, genetically mapped markers covering the genome (12–14) has enabled the development of a multistage paradigm for mapping genes for complex traits. In the first stages, complete genome screening (e.g., through lod score analysis) is used to identify possible localizations for disease genes. Subsequently, the regions highlighted by the screening study are more intensively investigated to confirm the initial localizations and delineate clear candidate regions. Finally, fine mapping methods [such as haplotype or linkage disequilibrium (LD) analysis] or candidate gene approaches are used for positional cloning of disease genes. Such multistage strategies have recently been used to map (15–21) and clone (22) genes for complex nonpsychiatric disorders despite the absence of standard linkage data meeting traditional criteria for significance.
Our genome screening study for BP employed the strategies used to map genes for complex nonpsychiatric diseases (6). Unlike previous genetic studies of BP, this study considered as affected only those individuals with the most severe and clinically distinctive forms of BP (BP-I and schizoaffective disorder, manic type, SAD-M), rather than including those diagnosed with a milder form of BP (BP-II) or with unipolar major depressive disorder (MDD). We studied two large pedigrees (CR001 and CR004) from a genetically homogeneous population, that of the Central Valley of Costa Rica (23, 24). We screened the entire genome for linkage by using mapped microsatellite markers and a model for genetic analysis in which most of the linkage information derived from affected individuals. The goal of our stringent linkage analysis was to identify all regions potentially harboring major genes for BP-I in the study population, rather than to establish definitive linkage. We derived predesignated lod score thresholds (using linkage simulation analyses), to suggest regions worthy of further investigation. Importantly, although we anticipate observing LD in the immediate vicinity of a disease gene among affected individuals in a population (such as that of the Central Valley) with a small number of founders, this expectation does not invalidate the assumption usually made for standard lod score analysis that markers are in linkage equilibrium with each other—i.e., their alleles are not associated. This is because the 10- to 20-cM chromosomal regions over which one is searching for evidence of linkage with the lod score method (i.e., cosegregation of marker and trait loci) are larger than those over which one expects to observe LD (i.e., allelic association) even in such a population (11, 19).
We performed the genome screening in two stages. The goals of the stage I screen were to identify areas suggestive of linkage, so that we could saturate them with available markers, and to pinpoint regions, referred to as “coverage gaps,” where markers were insufficiently informative in our sample to detect evidence of linkage. The goal of the stage II screen was to follow up on regions flanking each marker that yielded peak lod scores approximately equal to or greater than the thresholds used for the coverage calculations, which were deemed regions of interest, and to fill in coverage gaps. We report here the results of our complete genome screening (stages I and II) using 473 markers.
MATERIALS AND METHODS
Pedigrees.
We studied two independently ascertained Costa Rican pedigrees (CR001 and CR004) that were chosen because they contained a high density of individuals with BP-I and because their ancestry could be traced to the founding population of the Central Valley of Costa Rica (24). The current population of the Central Valley (consisting of about two million people) is predominantly descended from a small number of Spanish and Amerindian founders in the 16th and 17th centuries (23). Studies of several inherited diseases have confirmed the genetic isolation of this population (25, 26). An extensive description of pedigrees CR001 and CR004 is provided in a separate paper (24). In the course of the study we discovered two links between these pedigrees (one occurred several generations ago and one occurred in a recent generation). We analyzed the families separately, however, because these links were discovered after the simulation analyses were completed and after the genome screening study had been initiated.
All available adult members of these families were interviewed in Spanish, using the Schedule for Affective Disorders and Schizophrenia Lifetime version (SADS-L) (27). Individuals who received a psychiatric diagnosis were interviewed again in Spanish by a research psychiatrist using the Diagnostic Interview for Genetic Studies (DIGS) (28). The interviews and medical records were then reviewed by two blinded best estimators who reached a consensus diagnosis. The diagnostic procedures are described in detail in ref. 24.
Genotyping Studies.
We used linkage simulations to select the most informative individuals from pedigrees CR001 and CR004 for genotyping studies (24). Under a 90% dominant model, simulation analyses with these individuals suggested that we would likely detect evidence of linkage (e.g., a probability of 92% of obtaining lod > 1.0 in the combined data set), using markers with an average heterozygosity of 0.75 spaced at 10-cM intervals (as discussed in ref. 24). For the stage I screen, we chose the most polymorphic markers (307 in total) placed at approximately 10-cM intervals on the 1992 Genethon map (11). These markers were then supplemented by a small number of markers from the Cooperative Human Linkage Center (CHLC) public data base. For the stage II screen we added 166 markers from newer Genethon and CHLC maps as they became available (13, 14) and from the public data base of the Utah Center for Genome Research. Genotyping procedures were as described previously (29). Briefly, one of the two PCR primers was labeled radioactively by using a polynucleotide kinase, and PCR products were separated by electrophoresis on polyacrylamide gels. Autoradiographs were scored independently by two raters. Data for each marker were entered into the computer data base twice, and the resultant files were compared for discrepancies.
Statistical Analyses.
Two-point linkage analyses were performed for all markers. Marker allele frequencies were estimated from the combined data set with correction for dependency due to family relationships (30). The linkage analyses included the 65 individuals who were genotyped as well as an additional 65 individuals who had been diagnostically evaluated but not genotyped. Only individuals with BP-I were considered affected, with the exception of two persons, one in each family, who carry diagnoses of SAD-M. The SAD-M individuals were included as affected because BP-I and SAD-M co-segregate in families and they are often difficult to distinguish from each other on the basis of their clinical presentation and course of illness (1, 2, 24). In all, we designated as affected 20 individuals within CR004 (16 available for genotyping) and 10 individuals from CR001 (8 available for genotyping). The phenotype for all other individuals was considered unknown except for 17 individuals who were designated as unaffected if they carried no psychiatric diagnoses and were well beyond the age of risk for BP-I (these individuals contributed little information to the linkage analysis).
Linkage analyses were performed using a nearly dominant model (assuming penetrance of 0.81 for heterozygous individuals and 0.9 for homozygotes with the disease mutation). This model was chosen from five different single-locus models (ranging from recessive to nearly dominant) because it seemed most consistent with the segregation patterns of BP in the two pedigrees and because it had demonstrated the greatest power to detect linkage in simulation studies (24). On the basis of Costa Rican epidemiological surveys (23), we assumed the population prevalence of BP-I to be 0.015 (and thus the frequency of the disease allele to be 0.003). The frequency of BP-I in individuals without the disease allele was set at 0.01, which effectively specified a population phenocopy rate of 0.67 (i.e., an affected individual in the general population has a 2/3 probability of being a phenocopy). For multiply affected families, the probability that a gene is segregating is highly increased, which implies that affected individuals in our study pedigree have a lower probability to be phenocopies than affected individuals in the general population, particularly those with several affected close relatives (the exact probabilities are dependent on the degree of relationship between patients and the number of intervening affected individuals). These parameters were chosen to ensure that most of the linkage information derives from affected individuals. The rationale for selecting these parameters and results of analyses that demonstrate the conservatism of this model have been reported elsewhere (24). Due to the uncertainties regarding the parameters of genetic transmission, we did not perform multipoint linkage analysis (31).
Coverage.
To assess coverage for a marker, we calculated the number of informative meioses at the estimated recombination fraction, using the estimate of the variance (the inverse of the information matrix) (32). Alternatively, when the estimated frequency of recombination was close to 0 or 1, we applied Edwards’ equation to calculate the equivalent number of observations (33). These meioses represent the amount of linkage information provided by the marker, given the pedigree structure and the genetic model applied. We then assumed linkage to the marker in question and calculated the lod score that would be observed as a disease gene is hypothetically moved in increments away from that marker. All regions around a marker that would have generated a lod score that exceeded our predesignated thresholds for possible linkage (0.8 in CR001, 1.2 in CR004, and 1.6 in the combined data) were considered covered. These lod score thresholds were derived from simulation analyses showing the expected distribution of lod scores under linkage and nonlinkage (24), and they approximately represent a result that is 250 times more likely to occur in linked simulations than in unlinked simulations. We constructed coverage maps (Fig. 1) by superimposing the regions covered by each marker on the genetic map of each chromosome. At the end of the stage II screening we had typed a total of 473 microsatellite markers, with genome coverage (in the combined data set) of over 94%. Possible coverage gaps are indicated by unshaded areas and are mainly concentrated near telomeres. Because the coverage calculations make use of marker informativeness within the pedigrees, the coverage approach thus permits us to detect instances where markers with expected high heterozygosities are uninformative in our data set.
Figure 1.
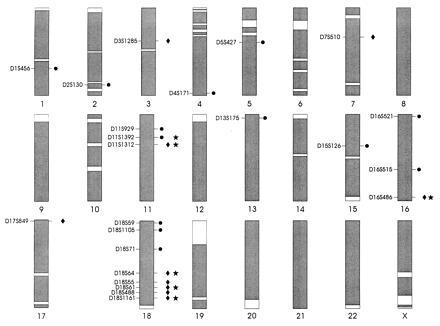
Extent of marker coverage of each chromosome. Coverage is defined as regions for which a lod score of at least 1.6 would have been detected (in the combined data set) for markers truly linked to BP-I under the model that we employed. Areas that remain uncovered (at this threshold) are unshaded. Markers for which lod scores were obtained that exceeded the predesignated coverage thresholds in CR001, CR004, or the combined data set are shown at their approximate chromosomal position. The symbols to the right of the chromosome indicate the thresholds exceeded at that marker: • signifies that the lod score at a marker exceeded the threshold of 0.8 in CR001; ♦ signifies that the lod score exceeded the threshold of 1.2 in CR004; and ★ signifies that the lod score exceeded the threshold of 1.6 in the combined data set.
RESULTS
Linkage Analysis.
Of the 473 microsatellites analyzed with two-point linkage tests, 23 markers exceeded the predesignated thresholds designated for the coverage calculations (in either CR001 or CR004, or in the combined data set). The location of these markers, the peak lod scores obtained in each family and in the combined data set, and the maximum likelihood estimate of the recombination fraction (θ) at which these lod scores were observed are indicated in Table 1. The approximate chromosomal locations of these markers are also depicted in Fig. 1. The distribution of lod scores (for the maximum likelihood estimate of θ in the combined data set) across the genome is displayed by chromosome in Fig. 2.
Table 1.
Lod scores for markers exceeding the predesignated coverage thresholds
| Marker name | Distance from pter, cM | Family CR001
|
Family
CR004
|
Combined
|
|||
|---|---|---|---|---|---|---|---|
| Zmax ≥ 0.8 | θ | Zmax ≥ 1.2 | θ | Zmed ≥ 1.6 | θ | ||
| D1S456 | 224.6 | 1.32 | 0.0 | 0.0 | 0.50 | 0.0 | 0.50 |
| D2S130 | 230.1 | 0.89 | 0.0 | 0.12 | 0.35 | 0.36 | 0.26 |
| D3S1285 | 91.0 | 0.00 | 0.50 | 2.59 | 0.00 | 1.15 | 0.16 |
| D4S171 | 207.9 | 1.07 | 0.07 | 0.01 | 0.05 | 0.22 | 0.29 |
| D5S427 | 69.6 | 1.39 | 0.0 | 0.0 | 0.50 | 0.7 | 0.18 |
| D7S510 | 60.5 | 0.04 | 0.40 | 2.04 | 0.0 | 0.82 | 0.17 |
| D11S929 | 36.3 | 0.80 | 0.11 | 0.03 | 0.42 | 0.43 | 0.24 |
| D11S1392 | 38.6 | 0.86 | 0.07 | 0.90 | 0.23 | 1.58 | 0.19 |
| D11S1312 | 42.0 | 0.47 | 0.13 | 1.77 | 0.0 | 1.95 | 0.05 |
| D13S175 | 7.4 | 0.83 | 0.0 | 0.0 | 0.50 | 0.24 | 0.15 |
| D15S126 | 45.5 | 1.09 | 0.0 | 0.0 | 0.48 | 0.06 | 0.40 |
| D16S521 | 4.6 | 1.46 | 0.0 | 0.41 | 0.26 | 1.18 | 0.17 |
| D16S515 | 94.8 | 0.93 | 0.09 | 0.01 | 0.46 | 0.39 | 0.25 |
| D16S486 | 133.6 | 0.27 | 0.19 | 1.29 | 0.20 | 1.60 | 0.20 |
| D17S849 | 0.60 | 0.0 | 0.50 | 1.22 | 0.07 | 0.32 | 0.14 |
| D18S59 | 1.1 | 1.43 | 0.0 | 0.0 | 0.50 | 0.02 | 0.46 |
| D18S1105 | 2.8 | 0.97 | 0.0 | 0.01 | 0.47 | 0.01 | 0.46 |
| D18S71 | 43.8 | 0.96 | 0.0 | 0.0 | 0.50 | 0.0 | 0.50 |
| D18S64 | 84.0 | 0.33 | 0.11 | 1.34 | 0.15 | 1.67 | 0.13 |
| D18S55 | 95.5 | 0.0 | 0.50 | 2.09 | 0.13 | 1.51 | 0.18 |
| D18S61 | 103.8 | 0.0 | 0.50 | 2.26 | 0.12 | 1.94 | 0.16 |
| D18S488 | 105.6 | 0.0 | 0.50 | 1.26 | 0.14 | 1.02 | 0.19 |
| D18S1161 | 113.0 | 0.0 | 0.50 | 1.79 | 0.16 | 1.76 | 0.17 |
Markers for which lod scores exceeded the predesignated thresholds used for genome coverage calculations are in boldface type. Zmax is the maximum likelihood estimate of the lod score at the corresponding value of the recombination fraction (θ).
Figure 2.
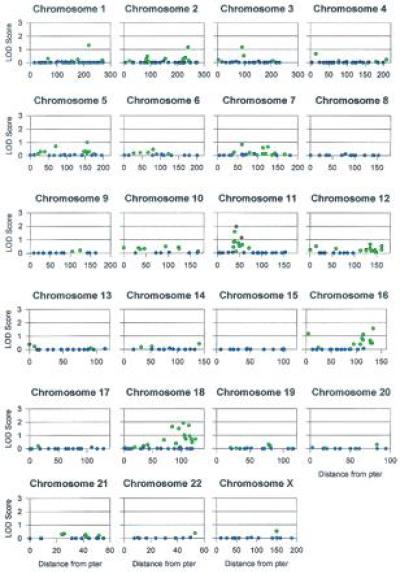
The lod scores for the maximum likelihood estimates (MLEs) of θ in the combined sample for the 473 microsatellite markers typed in the genome screen. The MLEs of θ are represented by different colors: red, θ < 0.10; green, 0.10 ≤ θ < 0.40; blue, θ ≥ 0.40. Note that the scale for the x-axis (distance from pter in cM) changes with chromosomes. Marker names and actual MLEs of θ are available from N.B.F. by request.
The region most suggestive of linkage to BP-I is 18q22–q23, where five markers exceeded the lod score thresholds in pedigree CR004, including three of the six markers (over the whole genome) that exceeded the threshold for the combined data set. (Detailed investigation of this region is described in ref. 34). The next most promising region is 11p13–p14, where lod scores at two contiguous markers equaled or exceeded the threshold for the combined data sets and a third adjacent marker exceeded the threshold in family CR001. The threshold for the combined data set was also equaled for a single marker near the telomere of 16q (D16S486). The threshold was exceeded for pedigree CR001 in two adjacent markers near the 18p telomere, but CR004 displayed no suggestion of linkage in this region. Finally, two of the highest lod scores were observed in CR004 at markers D3S1285 (2.59) and D7S510 (2.04), both peaks occurring at θ = 0; however, lod scores for markers that flank these two loci were positive but did not exceed any of the predesignated thresholds.
The cluster of suggestive lod scores in 18q22–q23 is adjacent to, but does not appear to overlap with, a possible localization to 18q21 for BP (BP-I and BP-II together) recently reported in a large set of small American families (35). Otherwise, the promising regions that we identified are not near possible localizations for broadly defined BP that have been proposed on chromosomes 11p15 (7), 21q22 (36), Xq28 (8), and in the pericentromeric region of 18 (37) or for BP-I in Xq24–q27 (38).
DISCUSSION
The genome screening study presented in this paper represents the first stage of our effort to identify all genes that play a major role in determining susceptibility to BP-I in the genetically homogeneous population of the Central Valley of Costa Rica. Our aim in this study was to detect those regions that merit further investigation, rather than to declare unequivocal linkage to a particular marker or markers. We used a conservative approach to linkage analysis in that almost all of the information for linkage is derived from individuals with a severe, narrowly defined phenotype. While this approach made it very unlikely that we would obtain lod scores greater than conventional thresholds of statistical significance (e.g., ≥ 3), it increased our confidence in the robustness of the most suggestive findings.
Our goal of identifying all suggestive regions and weighing the relative importance of findings required complete screening of the genome. We developed the coverage approach to gauge the progress of this effort. Conventionally, the thoroughness of genome screening is evaluated by excluding genome regions from linkage under given genetic models. This approach, which is highly sensitive to misspecification of genetic models, may be poorly suited for genome screening studies of complex traits; it is tied to the expectation of finding linkage at a single locus and demonstrating absence of linkage at all other locations in the genome. Additionally, exclusion analyses do not differentiate between genome regions where linkage is not excluded because markers are uninformative in the study population from those in which the genotype data are simply ambiguous. In contrast, the coverage approach is designed for studies aimed at genome screening rather than for studies whose goal is to demonstrate a single unequivocal linkage finding, and it provides explicit data regarding the informativeness of markers in the study pedigrees. Its use lessens the possibility that one would prematurely dismiss a given genome region as being unpromising for further study.
Because the exact genetic length of chromosomes is not clearly established, it is impossible to be certain that one has screened the entire genome. Although we report that we have covered about 94% of the genome (under the 90% dominant model) at the thresholds described above, we believe that this probably represents an underestimate. The remaining coverage gaps in our study occur predominantly at or near telomeres; as we used the upper-bound estimates for the length of each chromosome, it is likely that the actual coverage gaps in these regions are smaller than our conservative assessment.
Coverage results are model dependent and, given the uncertainty regarding the true model of genetic transmission of BP-I, it is possible that other potential localizations would emerge under different models. We therefore plan to reanalyze our data under a range of single-locus models (24).
In weighing the relative importance of our findings, we considered the presence of consistently positive lod scores over a given region to be of greater significance than isolated peak lod scores. Such clustering suggests true cosegregation of markers and phenotypes (i.e., alleles are shared identical by descent rather than identical by state) and is more readily observed in analyses of a few large pedigrees (as in our study) than in examinations of several smaller families. The most striking clustering of suggestive results is found in 18q22–q23. The localization of a BP-I gene in this region is supported by more extensive studies, as discussed in a previous paper (34). Other regions that merit further investigation on the basis of clustering of positive lod scores include 11p13–p14 and the telomere of 18p. Although we recognize that isolated occurrences of suggestive lod scores are likely to represent spurious results, the magnitudes of the lod scores obtained in CR004 at markers D3S1285 and D7S510 render these regions worthy of further examination.
In conclusion, through genome screening (the first stage of a multistep process for identifying genes for complex traits), we have identified regions that warrant further study in the search for BP-I genes. The second and third steps in this process consist of delineating clear candidate regions and fine mapping studies. The genetic homogeneity of the Costa Rican population should facilitate our completion of these steps; it should be feasible to collect and genotype an additional independent sample of BP-I patients from the Costa Rican Central Valley to conduct haplotype and LD analyses. It is thus practical to positionally clone one or more genes that play a role in BP-I susceptibility in this population.
Acknowledgments
We thank S. Blower, V. Carlton, and L. Bull for helpful comments and N. Stricker and H. Consengco for technical assistance. Above all, we thank the members of families CR001 and CR004, and the Costa Rican institutions that made this work possible: the Hospital Nacional Psiquiatrico, the Hospital Calderon Guardia, the Ministry of Science, and the Caja Costamicense de Seguro Social. This work was supported by the National Institutes of Health (Grants MH00916, MH49499, MH48695, and MH47563), a Veterans Administration Research Psychiatrist Award (to L.A.M.), a Young Investigator Award from the National Alliance for Research on Schizophrenia and Depression (to N.B.F.), and a grant from the American Psychiatric Association’s Program for Minority Research Training in Psychiatry (to M.A.E.).
Footnotes
Abbreviations: BP, bipolar mood disorder; cM, centimorgan(s); LD, linkage disequilibrium; MDD, major depressive disorder; SAD-M, schizoaffective disorder, manic type.
References
- 1.Goodwin F K, Jamison K R. Manic Depressive Illness. New York: Oxford Univ. Press; 1990. pp. 373–401. [Google Scholar]
- 2.Freimer N B, Reus V I. In: The Molecular and Genetic Basis of Neurological Disease. Rosenberg R N, Prusiner S B, DiMauro S, Barchi R L, Kunkel L, editors. Boston: Butterworth; 1993. pp. 951–965. [Google Scholar]
- 3.Bertelsen A, Harvald B, Hauge M. Br J Psychiatry. 1977;130:330–351. doi: 10.1192/bjp.130.4.330. [DOI] [PubMed] [Google Scholar]
- 4.Pauls D L, Bailey J N, Carter A S, Allen C R, Egeland J A. Am J Med Genet. 1995;60:290–297. doi: 10.1002/ajmg.1320600406. [DOI] [PubMed] [Google Scholar]
- 5.Spence M A, Kladman P L, Sadovnick A D, Bailey-Wilson J E, Ameli H, Remick R A. Am J Med Genet. 1995;60:370–376. doi: 10.1002/ajmg.1320600505. [DOI] [PubMed] [Google Scholar]
- 6.McInnes L A, Freimer N B. Curr Opin Genet Dev. 1995;5:376–381. doi: 10.1016/0959-437x(95)80054-9. [DOI] [PubMed] [Google Scholar]
- 7.Egeland J A, Gerhard D S, Pauls D L, Sussex J N, Kidd K K, Allen C R, Hostetter A M, Housman D E. Nature (London) 1987;325:783–787. doi: 10.1038/325783a0. [DOI] [PubMed] [Google Scholar]
- 8.Baron M, Risch N, Hamburger R, Mandel B, Kushner S, Newman M, Drumer D, Belmaker R H. Nature (London) 1987;326:289–292. doi: 10.1038/326289a0. [DOI] [PubMed] [Google Scholar]
- 9.Kelsoe J R, Ginns E I, Egeland J A, Gerhard D S, Goldstein A M, Bale S J, Pauls D L, Long R T, Kidd K K, Conte G, Housman D E, Paul S M. Nature (London) 1989;342:238–243. doi: 10.1038/342238a0. [DOI] [PubMed] [Google Scholar]
- 10.Baron M, Freimer N, Risch N, Lerer B, Alexander J R, Straub R E, Asokan S, Das K, Peterson A, Amos J, Endicott J, Ott J, Gilliam T C. Nat Genet. 1993;3:49–55. doi: 10.1038/ng0193-49. [DOI] [PubMed] [Google Scholar]
- 11.Houwen R H, Baharloo S, Blankenship K, Raeymaekers P, Juyn J, Sandkuijl L A, Freimer N B. Nat Genet. 1994;8:380–386. doi: 10.1038/ng1294-380. [DOI] [PubMed] [Google Scholar]
- 12.Weissenbach J, Gyapay G, Dib C, Vignal A, Morissette J, Millasseau P, Vaysseix G, Lathrop M. Nature (London) 1992;359:794–801. doi: 10.1038/359794a0. [DOI] [PubMed] [Google Scholar]
- 13.Murray J C, Buetow K H, Weber J L, Ludwigsen S, Scherphier-Heddema T, Manium F, Quillen J, Sheffield V C, Sundin S, Duyk G M. Science. 1994;265:2049–2054. doi: 10.1126/science.8091227. [DOI] [PubMed] [Google Scholar]
- 14.Gyapay G, Morissette J, Vignal A, Dib C, Fizanes C, Millasseau P, Marc S, Bernardi G, Lathrop M, Weissenbach J. Nat Genet. 1994;7:246–339. doi: 10.1038/ng0694supp-246. [DOI] [PubMed] [Google Scholar]
- 15.Davies J L, Kawaguchi Y, Bennett S T, Copeman J B, Cordell H J, Pritchard L E, Reed P W, Gough S C L, Jenkins S C, Palmer S M, Balfour K M, Rowe B R, Farrall M, Barnett A H. Nature (London) 1994;371:130–136. doi: 10.1038/371130a0. [DOI] [PubMed] [Google Scholar]
- 16.Hashimoto L, Habita C, Beressi J P, Delepine M, Besse C, Cambon-Thomsen A, Deschamps I, Rotter J I, Djoulag S, James M R, Froguel P, Weissenbach J, Lathrop G M, Julier C. Nature (London) 1994;371:161–164. doi: 10.1038/371161a0. [DOI] [PubMed] [Google Scholar]
- 17.Copeman J B, Cucca F, Hearne C M, Cornall R J, Reed P W, Ronningen K S, Undlien D E, Nistico L, Buzzetti R, Tosi F, Pociot F, Nerup J, Cornelis F, Barnett A H, Todd J A. Nat Genet. 1995;9:80–85. doi: 10.1038/ng0195-80. [DOI] [PubMed] [Google Scholar]
- 18.Puffenberger E G, Kauffman E R, Bolk S, Matise T C, Washington S S, Angrist M, Weissenbach J, Garver K L, Mascari M, Ladda R, Slaugenhaupt S A, Chakravarti A. Hum Mol Genet. 1994;3:1217–1225. doi: 10.1093/hmg/3.8.1217. [DOI] [PubMed] [Google Scholar]
- 19.Puffenberger E G, Lynn A, Kunst C, Warren S, Chakravarti A. Am J Hum Genet. 1995;57:A42. [Google Scholar]
- 20.Ozelius L J, Kramer P L, de Leon D, Risch N, Bressman S B, Schuback D E, Brin M F, Kwiatkowski D J, Burke R E, Gusella J F, Fahn S, Breakefield X O. Am J Hum Genet. 1992;50:619–628. [PMC free article] [PubMed] [Google Scholar]
- 21.Friedman T B, Liang Y, Weber J L, Hinnant J T, Barber T D, Winala S, Arkya I N, Asher J H. Nat Genet. 1995;9:86–91. doi: 10.1038/ng0195-86. [DOI] [PubMed] [Google Scholar]
- 22.Puffenberger E G, Hosoda K, Washington S S, Nakao K, deWit D, Yanagisawa M, Chakravarti A. Cell. 1994;79:1257–1266. doi: 10.1016/0092-8674(94)90016-7. [DOI] [PubMed] [Google Scholar]
- 23.Escamilla M A, Spesny M, Reus V I, Gallegos A, Meza L, Moina J, Sandkuijl L A, Fournier E, Leon P A, Smith L B, Freimer N B. Am J Med Genet. 1996;67:244–253. doi: 10.1002/(SICI)1096-8628(19960531)67:3<244::AID-AJMG2>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 24.Freimer N B, Reus V I, Escamilla M A, Spesny M, Smith L B, Service S K, Gallegos A, Meza L, Batki S, Vinogradov S, Leon P, Sandkuijl L A. Am J Med Genet. 1996;67:254–263. doi: 10.1002/(SICI)1096-8628(19960531)67:3<254::AID-AJMG3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 25.Leon P, Raventos H, Lynch E, Morrow J, King M C. Proc Natl Acad Sci USA. 1992;89:5181–5184. doi: 10.1073/pnas.89.11.5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uhrhammer N, Lange E, Porras O, Naeim A, Chen X, Sheikhavandi S, Chiplunkar S, Yang L, Dandekar S, Liag T, Patel N, Teraoka S, Udar N, Calvo N, Concannon P, Lange K, Gatti R A. Am J Hum Genet. 1992;57:103–111. [PMC free article] [PubMed] [Google Scholar]
- 27.Endicott J, Spitzer R L. Arch Gen Psychiatry. 1978;35:837–844. doi: 10.1001/archpsyc.1978.01770310043002. [DOI] [PubMed] [Google Scholar]
- 28.Nurnberger J I, Blehar M C, Kaufmann C A, York-Cooler C, Simpson S G, Harkavy-Friedman J, Severe J B, Malaspina D, Reich T. Arch Gen Psychiatry. 1994;51:849–859. doi: 10.1001/archpsyc.1994.03950110009002. [DOI] [PubMed] [Google Scholar]
- 29.DiRienzo A, Peterson A C, Garza J C, Valdes A M, Slatkin M, Freimer N B. Proc Natl Acad Sci USA. 1994;91:3166–3170. doi: 10.1073/pnas.91.8.3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boehnke M. Am J Hum Genet. 1991;48:22–25. [PMC free article] [PubMed] [Google Scholar]
- 31.Risch N, Giuffra L. Hum Hered. 1992;42:77–92. doi: 10.1159/000154047. [DOI] [PubMed] [Google Scholar]
- 32.Petrukhin K E, Speer M C, Cayamis E, Bonaldo M F, Tantravahi U, Sares M B, Fischer S G, Warburton D, Gilliam T C, Ott J. Genomics. 1993;15:76–85. doi: 10.1006/geno.1993.1012. [DOI] [PubMed] [Google Scholar]
- 33.Edwards J H. Ann Hum Genet. 1971;34:229–250. doi: 10.1111/j.1469-1809.1971.tb00237.x. [DOI] [PubMed] [Google Scholar]
- 34.Freimer N B, Reus V I, Escamilla M A, McInnes L A, Spesny M, Leon P, Service S K, Smith L B, Silva S, Rojas E, Gallegos A, Meza L, Fournier E, Baharloo S, Blankenship K, Tyler D J, Batki S, Vinogradov S, Weissenbach J, Barondes S H, Sandkuijl L A. Nat Genet. 1996;12:236–241. doi: 10.1038/ng0496-436. [DOI] [PubMed] [Google Scholar]
- 35.Stine O C, Su J, Koskela R, McMahon F J, Gschwend M, Friddle C, Clark C C, McInnis M G, Simpson S G, Breschel T S, Vishio E, Riskin K, Feilotter H, Chen E, Shen S, Folstein S, Meyers D A, Botstein D, Marr T G, DePaulo J R. Am J Hum Genet. 1995;56:1384–1394. [PMC free article] [PubMed] [Google Scholar]
- 36.Straub R E, Lehner T, Luo Y, Loth J E, Shao W, Sharpe L, Alexander J R, Das K, Simon R, Fieve R R, Lerer B, Endicott J, Ott J, Gilliam T C, Baron M. Nat Genet. 1994;8:291–296. doi: 10.1038/ng1194-291. [DOI] [PubMed] [Google Scholar]
- 37.Berrettini W H, Ferraro T N, Goldin L R, Weeks D E, Detera-Wadleigh S, Nurnberger J I, Gershon E S. Proc Natl Acad Sci USA. 1994;91:5918–5921. doi: 10.1073/pnas.91.13.5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pekkarinen P, Terwilliger J, Bredbacka P, Lonnquist J, Peltonen L. Genome Res. 1995;5:105–115. doi: 10.1101/gr.5.2.105. [DOI] [PubMed] [Google Scholar]