

Abstract
The isolation and study of Anopheles gambiae genes that are differentially expressed in development, notably in tissues associated with the maturation and transmission of the malaria parasite, is important for the elucidation of basic molecular mechanisms underlying vector–parasite interactions. We have used the differential display technique to screen for mRNAs specifically expressed in adult males, females, and midgut tissues of blood-fed and unfed females. We also screened for mRNAs specifically induced upon bacterial infection of larval stage mosquitoes. We have characterized 19 distinct cDNAs, most of which show developmentally regulated expression specificity during the mosquito life cycle. The most interesting are six new sequences that are midgut-specific in the adult, three of which are also modulated by blood-feeding. The gut-specific sequences encode a maltase, a V-ATPase subunit, a GTP binding protein, two different lectins, and a nontrypsin serine protease. The latter sequence is also induced in larvae subjected to bacterial challenge. With the exception of a mitochondrial DNA fragment, the other 18 sequences constitute expressed genomic sequence tags, 4 of which have been mapped cytogenetically.
Blood feeding by female mosquitoes provides the means of transmission for a variety of viral, protozoan, and metazoan disease agents. Plasmodium, the protozoon causing malaria, enters the mosquito midgut with the blood meal where it undergoes gametogenesis and fertilization, before developing into an ameboid cell (ookinete) that must traverse the epithelial cells. In the midgut the developing parasite must resist attack by immunoreactive molecules and digestive enzymes, penetrate through the chitinous peritrophic membrane, and cross the gut wall to reach the basal lamina (1). There it multiplies vegetatively as an oocyst, unless it is blocked by melanotic encapsulation of unknown origin, as occurs in certain genetic strains of mosquitoes selected for refractoriness to the parasite (2). Thus, the midgut constitutes an important potential barrier to malaria transmission.
We are studying the molecular genetics of the mosquito, Anopheles gambiae, one of the most important disease vectors in sub-Saharan Africa. Identification of the genes expressed in the digestive tract of the mosquito is of obvious importance for elucidation of the mechanisms that permit parasite development and successful passage through the midgut wall. However, only a few midgut-specific genes have been isolated to date, largely those encoding digestive proteases (3). Other components involved in the development and physiology of the midgut, as well as immune effectors which might operate in the midgut environment, remain to be identified and studied. Promoters of midgut-specific genes are also of potential interest, because they may be used in the future for engineering new types of malaria-refractory mosquitoes, specifically expressing Plasmodium-blocking agents (4). More broadly, isolation of differentially expressed mosquito genes should provide tools for analyzing development and physiology, potentially contributing to our understanding of how the vector interacts with insect-specific stages of the malaria parasite.
Until recently, the methods of choice for identification of differentially expressed genes were construction of subtracted cDNA libraries, or differential screening of ordinary genomic or cDNA libraries. In both cases, the availability of sufficient starting material can represent a problem, as is often true for mosquito studies. This problem has been circumvented by the use of PCR-based techniques that use primers of arbitrary sequence to amplify, and subsequently isolate, differentially expressed mRNA sequences. An adaptation of the random amplified polymorphic DNA method of detecting DNA polymorphisms (5) uses a single arbitrary primer for both reverse transcription (RT) and the amplification of cDNAs (6). In a second method, oligo(dT) with or without a partial anchor (7) is used to prime cDNAs, which are then amplified using an arbitrary decanucleotide as the 5′ primer. In both cases, the radioactively labeled amplification products are resolved by electrophoresis on sequencing gels. In such “differential display” methods, comparison of samples processed in parallel permits rapid and easy identification of a set of candidate tissue-specific or developmental stage-specific gene products. Although these methods were developed using abundantly available biological material, the use of PCR amplification can allow identification of even rare mRNA sequences in tissues that are difficult to obtain.
In the present study we used a streamlined differential display technique, coupled with PCR-based verification assays, to isolate mosquito cDNA sequences potentially expressed in a differential manner. Six selected sequences are preferentially expressed in the adult midgut, our primary target organ. We have isolated two additional sequences that are affected by blood feeding. Most of the sequences that were examined carefully show some stage specificity, while only four represent constitutive or housekeeping components, one is female specific, and one is induced by bacterial infection of the larvae.
MATERIALS AND METHODS
Mosquito Colony and Infections.
An. gambiae G3 and Suakoko strains were reared at 28°C, 75% humidity, with a 12-h light/dark cycle. Adults were maintained on 10% sucrose solution and females were blood-fed on anesthetized rats. Larvae were experimentally infected by pricking with a needle dipped in a bacterial pellet of Escherichia coli (strain 1106) and Micrococcus luteus (strain A270) and allowed to recover for varying time periods in clean H20.
RNA Extraction and Differential Display.
RNA was prepared from midguts and remaining carcasses of adult female An. gambiae. The blood meal was removed from the fed guts prior to RNA extraction. Total RNAs were extracted from these samples or entire animals at the indicated stages by using the RNaid PLUS kit (Bio 101) according to the manufacturer’s instructions, followed by DNase I treatment.
Differential display, G and S series.
First-strand oligo(dT)-primed cDNA synthesis was performed in 50 μl reactions consisting of 1 μg RNA, 50 mM Tris·HCl (pH 8.3), 50 mM KCl, 4 mM MgC12, 0.1 mM each dNTP, 20 mM DTT, 6 μM oligo(dT) primer [5′-CGGCCTCGAC(T)12-3′], 20 units RNasin (Promega) and 40 units Moloney murine leukemia virus reverse transcriptase (Pharmacia). Reactions were incubated at 37°C for 90 min and then heated to 60°C for 10 min. The mix was centrifuged through a Sephadex G-25 column (Pharmacia) and adjusted to 100 μl. For PCR amplification, 2 μl of the cDNA template was used in 25 μl reactions containing 10 mM Tris·HCl (pH 8.3), 25 mM KCl, 2 mM MgCl2, 0.1 mM each dNTP, 0.5 μM decamer primer (Operon Technologies, Alameda, CA) and 0.5 μM oligo(dT) primer (as for first-strand cDNA synthesis). Forty-five amplification cycles (1 min at 94°C, 1 min at 40°C, 2 min at 72°C) were performed in a Perkin–Elmer thermal cycler. Reaction products were resolved on a 1.4% agarose gel, visualized by ethidium bromide staining, and excised for cloning.
Differential display, A series.
DNase I-treated total RNA (0.5 μg) was reverse transcribed using degenerate oligo(dT) primers designated T12MA, T12MG, T12MC, or T12MT (where M represents A, G, or C). First-strand cDNA synthesis reactions were performed in a volume of 50 μl and contained 0.5 μg RNA, 2.5 μM T12MN primer, 20 μM each dNTP (Pharmacia), 40 units RNasin (Boehringer Mannheim), 50 mM Tris·HCl (pH 8.3), 75 mM KCl, 3 mM MgCl2, 10 mM DTT, and 100 units Superscript II reverse transcriptase (GIBCO/BRL). After incubation at 45°C for 1 h, 2 μl of the cDNA was used as template for differential display PCR. Differential display PCRs and subsequent isolation of candidate cDNAs were performed as described by Liang and Pardee (7).
Cloning and Sequencing.
The differentially amplified bands was gel purified and cloned in the EcoRV site of plasmid pBluescript (Stratagene) or cloned using the TA Cloning Kit (Invitrogen). Sequencing was performed with the Sequenase Kit (United States Biochemical) according to the manufacturers instructions. To obtain a full-length A16 clone, we used the A16 fragment as probe to screen a cDNA library prepared in λZAPII (Stratagene) from bacteria-infected larvae (8). Full-length coding sequences of G13 and G20 were obtained by PCR amplification using template DNA prepared from a cDNA library. The vector T3 priming site upstream of the cDNA insert was used in combination with specific primers G13B and G20B. Amplification products were cloned with the TA Cloning Kit (Invitrogen). Sequence analysis used the Genetics Computer Group software (9), and data bases searches used the blast programs (10).
RNA Blot Analysis.
Twenty micrograms of total RNA per lane was electrophoresed on 1.4% formaldehyde agarose gels and transferred to nylon membranes (Hybond; Amersham). Probes were prepared using the Hi Prime Labelling Kit (Boehringer Mannheim). Hybridizations were performed at 65°C in 7% SDS/1% BSA/0.5 mM EDTA/0.5 M phosphate buffered hybridization solution (11). Following hybridization, the filters were washed under conditions of high stringency in 10% SDS/0.5 M phosphate buffer.
In Situ Hybridization to Polytene Chromosomes.
Selected clones were mapped on nurse cell polytene chromosomes as described by della Torre et al. (12). Hybridization of cDNA clone A16 to the division-specific chromosomal library was performed as described by Zheng et al. (13).
Expression Analysis by RT–PCR.
First strand cDNA was synthesized using the Moloney murine leukemia virus reverse transcriptase (GIBCO/BRL) and the supplied buffers. Synthesis was primed using oligo(dT) coupled to magnetic beads (Dynal, Oslo). Reactions were performed at 37°C for 1 h and terminated by heating to 95°C for 5 min. Beads were rinsed and resuspended in distilled water. One microliter of the cDNA template was used in 25 μl PCRs containing primer pairs specific for the various cDNAs (see below), 50 mM KCl, 10 mM Tris·HCl (pH 8.3), 2 mM MgCl2, 20 pmol each primer, 100 μM dNTPs (Pharmacia), and 2.5 units Taq polymerase. Appropriate annealing temperatures and cycle numbers were estimated empirically for each primer pair separately. Reaction products were visualized on 1.4% ethidium-stained agarose gels. Primers specific for ribosomal protein S7 (14) or actin (15) cDNAs were used as controls. The primers used for these experiments were as follows: G3A, 5′-ACCTCAAAGTACAGCTTGCC-3′ and G3B, 5′-CTACGTGAAGGTTGTCAAGG-3′; G9A, 5′-ATTAGGCAATAACTTATCCG-3′ and G9B, 5′-TGAACCATTTATTTTTTG-3′; G10A, 5′-CCAACTTCATGGTTGTCG-3′ and G10B, 5′-GGGAGAGGTTCAAAATAC-3′; G11A, 5′-ATAATGCGTTCTTCTTTCCC-3′ and G11B, 5′-GACTCCGTTTCAAAATCCAC-3′; G13A, 5′-GTCCTGGGGAGGTATTCC-3′ and G13B, 5′- AGCACTTCATTTGAAGCC-3′; G14A, 5′-CGTATCCACAGACCGATG-3′ and G14B, 5′-GTTCTTCAGCCCTTTCTG-3′; G18A, 5′-ACTACACAGGAGAGTAAACC-3′ and G18B, 5′-CAGTTTAAATTTATTGTTTC-3′; G20A, 5′-CCTGTCCAGAAGAAGTCC-3′ and G20B, 5′-TAGATGTGAATGACATGG-3′; S2A, 5′-CTTTTGTGCTGGGATAGC-3′ and S2B, 5′-CAACGAAAAAATTACTCC-3′; A16A, 5′-TGCATTGGCGCCTCAGTA-3′ and A16B, 5′-TCCGTCGTTATCCACGTT-3′; A34A, 5′-CATTGTGGTTGAGGCCAC3′ and A34B, 5′-AGTATCACAATCGGTTTC-3′; A41A, 5′-CTATCACGGTAAACGGC-3′ and A41B, 5′-ACCTTAGCCTTTGCATG-3′; A48A, 5′-GTGAACAAAAGTCCCAGC3′ and A48B, 5′-GGCCATGAGTGATGTTAC-3′; A51A, 5′-CCTGTTAGACAGATATCC-3′ and A51B, 5′-GCACTGGAATTCTAACTC-3′; A71A, 5′-GTTCATTTATTTCGAGAC-3′ and A71B, 5′-CGTTGCCTTCTATGGCC-3′; A72A, 5′-CACGTAAAGGATGCTC-3′ and A72B, 5′-AACGAAGCACTGAAGTG-3′; S7A, 5′-GGCGATCATCATCTACGTTGC-3′ and S7B, 5′-GTAGCTGCTGCAAACTTCGG-3′; ActA, 5′- CCCATGATGCGCAGTGGATA-3′ and ActB, 5′-GAGTTGGCATGAGTCAGCGC-3′.
RESULTS
Differential Display and Overview of the cDNAs.
To obtain cDNAs that are differentially expressed in adult midguts, and might be modulated by feeding, RNA preparations from guts both before and 5, 24, 30, or 50 h after a blood meal were compared with each other and to RNAs from the respective gut-free carcasses. Various combinations of cDNAs were compared by the differential display procedure, using a total of 30 decamer primers of which 10 proved fruitful. In a first step, oligo(dT)-primed cDNAs were synthesized from total RNAs, and aliquots were then compared pairwise by PCR using various decamers and an oligo(dT) primer. To simplify the procedure and avoid radioisotopes, the reaction products were analyzed on ethidium bromide-stained agarose gels, considerably facilitating the isolation and cloning of amplified products (amplicons). Fig. 1 shows typical comparisons, leading to the isolation of three cDNAs that are reproducibly enriched in the gut, G3, G13, and G14. A total of 20 candidate gut-specific products (series G) were isolated in this manner. Similar comparisons of cDNAs from entire adult males and females led to the isolation of four candidate sex-specific products (series S), of which S1 and S2 are shown in Fig. 1. S1, as a male-specific amplicon and hence not relevant to parasite transmission, was not subjected to cloning and sequence analysis. Finally, 11 candidate cDNA clones (series A) were isolated from an experiment of different design, comparing transcripts in control and bacteria-challenged larvae (see Materials and Methods).
Figure 1.
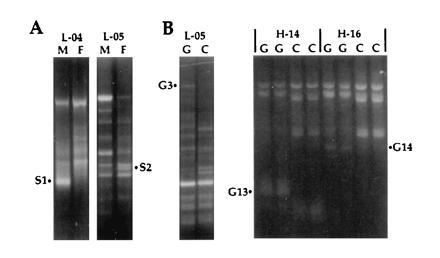
Examples of differential display comparing male versus female (A, M versus F) and gut versus carcass (B, G versus C) mRNA. Amplification products were resolved on 1.4% ethidium-stained agarose gels. PCR amplification used decamer primers L-04 (5′-GACTGCACAC-3′), L-05 (5′-ACGCAGGCAC-3′), H-14 (5′-ACCAGGTTGG-3′), and H-16 (5′-TCTCAGCTGG-3′), in combination with an oligo(dT) primer. H-14 and H-16 reactions were performed in duplicate. The differentially amplified PCR fragments S1, S2, G3, G13, and G14 were isolated for further analysis.
The 34 cloned fragments as well as the S1 amplicon were then used to probe RNA blots (Northern blot analysis), to confirm differential expression and estimate the sizes of the transcripts (Fig. 2A). Partial nucleotide sequences were determined, and the EMBL and SwissProt data bases were searched for similarities (10). Differential expression of the transcripts was tested with greater sensitivity by RT–PCR, using specific RNA preparations and clone-specific primers. At this stage, 15 clones were set aside because of inconclusive RT–PCR results, and the remaining 19 were analyzed with respect to differential expression. Fig. 2 and Table 1 summarize the results. Tissue- and stage-specific expression was evaluated by RT–PCR relative to known “housekeeping” transcripts, encoding the S7 ribosomal protein (14) and a cytoplasmic actin (15).
Figure 2.
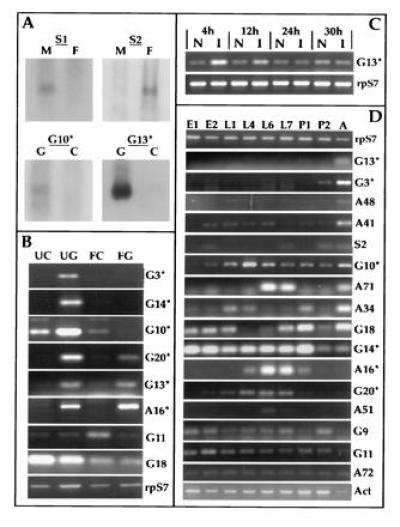
mRNA expression analysis of differentially amplified fragments. (A) Northern analysis of adult sex and gut-specific differential display fragments. The female-specific clone S2 as well as amplicon S1 were hybridized to filters containing RNA isolated from unfed females (F) and males (M). The gut-specific clones were hybridized to filters containing RNA from fed (24 h) midguts (G) and remaining carcasses (C). (B) RT–PCR analysis of gut specific and blood meal modulated expression, comparing unfed midguts (UG), carcasses (UC), blood-fed midguts (FG) and carcasses (FC) 22 h postfeeding. cDNA clones are identified on the right margin with asterisks, indicating those shown in Fig. 3. Levels of ribosomal protein S7 RNA were used as control. (C) RT–PCR analysis of G13 mRNA levels in bacterially challenged larvae versus naive larvae at 4, 12, 24, and 30 h following infection. (D) RT–PCR expression analysis comparing RNA from embryos (E1 = 0–24 h; E2 = 28–46 h), larvae (L1 = 0–25 h, L4 = 4 days, L6 = 6 days, L7 = 7 days), pupae (P1 = early and P2 = late), and adult females (A). Ribosomal protein S7 (15) and cytoskeletal actin (16) mRNA levels were used as controls.
Table 1.
Characterization of mosquito cDNAs
| Code | Accession no. | Sequence, bp | mRNA, kb | Cytology | Expression
pattern
|
Homology | % Identity (aa overlap) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Tissue and sex | Feeding | Immune challenge | Stage | |||||||
| G3 | Z69981Z69981 | 1001 | 1.9 | 44D | G | U | A > P2 | Maltase | 48 (326) | |
| G10 | Z69979Z69979 | 691* | 1.1 | 12B | G (C) | U (F) | L4, A > rest > E1 | V-ATPase | 90 (124) | |
| G13 | Z69978Z69978 | 1029* | 1.5 | G | U, F | I | A | Serine protease | 50 (266) | |
| G14 | Z69980Z69980 | 408 | 1.3 | 12B | G | U | E1, P1 > E2, L > P2, A | GTP binding | 97 (134) | |
| G20 | Z69982Z69982 | 612* | 0.8 | G | U, F | N | L4-L7 > L1 > E2, P1 | Gal-lectin | 26 (142) | |
| A16 | Y08163Y08163 | 672* | 0.7 | 20 | G | U, F | N | L6, L7 > L4, P1 > E2, P2 | C-lectin | 44 (45) |
| G11 | Z69954Z69954 | 379 | C | F | E2 > rest | |||||
| G18 | Z69960Z69960 | 150 | G, C | U (F) | A, P1 > E1, E2, L1, L7 > rest | |||||
| S2 | Z69975Z69975 | 418 | F | E2, L7, P2, A | ||||||
| G9 | Z69953Z69953 | 199 | E, L, P2 > P1 > A | |||||||
| A34 | Y08164Y08164 | 170 | N | A > L1, L4, P1 > rest | ||||||
| A41 | Y08165Y08165 | 146† | N | A > E2-L6 > P1, P2 | ||||||
| A48 | Y08166Y08166 | 191† | N | A > L1 | ||||||
| A51 | Y08167Y08167 | 206 | N | L6 > L7 | ||||||
| A71 | Y08168Y08168 | 119 | N | L6, L7, A > rest | ||||||
| A72 | Y08169Y08169 | 120 | N | Constitutive | ||||||
| G7 | Z69977Z69977 | 529 | Housekeeping | S11 | 68 (152) | |||||
| G5 | Z69976Z69976 | 754 | Housekeeping | L10 | 86 (203) | |||||
| S4 | 772 | Housekeeping | Mitochondrial | 100 | ||||||
Summary of available data on 19 cDNA clones identified by mRNA differential display PCR. Blank spaces indicate information not available. Sequence refers to the determined DNA sequence of each clone. ∗, Clones encompassing full-length coding sequences;
, partial sequence of larger cDNA fragments. mRNA transcript size is based on Northern blot analysis. Cytological location is based upon in situ hybridization to polytene chromosomes or chromosome division-specific DNA in the case of A16 (see Materials and Methods). RNA expression patterns were assayed by RT–PCR and/or Northern blot analysis. Sequences display tissue-specific expression with respect to gut (G), carcass (C), or adult female (F), show modulated expression as a consequence of blood-feeding by adult female mosquitoes (F = fed, U = unfed; 22 h following the bloodmeal), or were tested for immune induction of RNA as a consequence of bacterial challenge (I = induced expression in challenged larvae, N = no induction). Developmental stage-specific expression patterns shown in Fig. 2D are summarized here. Significant differences in expression are indicated in relative terms for each gene, from the developmental stage exhibiting highest levels to the lowest (see legend to Fig. 2D for designation of stages). The analysis was performed on the original photograph of the gels. Expression below the detection limit is possible at additional stages; this is examplified by G13, which is also detectable in larvae after additional PCR cycles (Fig. 2 C and D). Homologies were determined from blast analysis of nucleotide and/or deduced amino acid sequences. The % identity is based on the indicated overlap with the closest homologue, from which gaps have been excluded.
Midgut and Feeding-Specific cDNAs.
Comparison of transcripts from adult female gut and carcass identified certain sequences as unique to or highly enriched in the midgut. We focused special attention on six clones (Fig. 2B and Table 1) that show similarities to known sequences from other organisms (see Fig. 3 and Discussion). The corresponding genes were mapped by in situ hybridization to polytene chromosomes or by dot blot hybridization with chromosomal division-specific DNA pools (13). Comparisons between unfed and fed females (20 h after the blood meal) showed that all six of these sequences are expressed in the unfed gut. Three of them are switched off in the guts after feeding (G3, G10, G14), while three others (G13, G20, A16) continue to be detected or are even enhanced in the fed guts. Interestingly, one member of the latter category, G13, is also enhanced in bacteria-challenged larvae: it shows strong induction at 4 h postinfection, decreasing at 12 h and returning to normal levels by 24 and 30 h (Fig. 2C). Finally, G11 is enhanced in the fed carcass, while G18 is repressed by feeding, in both gut and carcass (Fig. 2B). Thus, a total of eight sequences are differentially expressed in the adult gut or are modulated by blood-feeding.
Figure 3.

Comparisons of deduced amino acid sequences of cloned cDNAs with previously characterized sequences from other organisms. Amino acid identities are indicated by bold type and asterisks. (A) An asterisk indicates the actual C terminus of the protein. First and/or last amino acid positions of the partial sequences are numbered in parentheses. The G3 sequence corresponding to the C-terminal part of a maltase-like protein is aligned with the An. gambiae proteins (17) corresponding to the maltase-like genes (line a) and 1 (line b). The conserved putative catalytic site (NHD) is boxed. (B) The full-length sequence of the putative C-type lectin A16 is aligned with the partial carbohydrate recognition domains of human l-selectin (line a) (18), human macrophage mannose receptor (line b) (19), and Sarcophaga lectin (line c) (20). Motifs characteristic of C-type lectins are boxed (ref. 21; A. Ezekowitz and N. Harris, personal communication). (C) The full-length sequence of the putative galactose lectin G20 is aligned with the rat intestinal lactose binding lectin (line a) (22), pig lactose binding lectin (line b) (23), and human Galectin-7 (line c) (24). Conserved amino acids involved in galactose binding are boxed. (D) The full-length sequence of the G10 putative 14K vacuolar ATPase is aligned with the homologous proteins of Drosophila melanogaster (line a) (accession no. S38436S38436), Manduca sexta (line b) (25); and human (line c) (accession no. JC4193). (E) The C-terminal part of the G14 putative low molecular weight G25K GTP binding protein is aligned with the homologous proteins of D. melanogaster (line a) (26) and human (line b) (27). (F) The full-length sequence of the G13 putative serine protease is aligned with Aedes aegypti chymotrypsin (line a) (accession no. U56423U56423), An. gambiae trypsin 7 (line b) (3), mouse mast cell protease 7 (line c) (28), and mouse tissue kallikrein (line d) (29). The residues (H, D, S) of the catalytic triad of serine proteases are underlined. The putative activation peptide (3) is marked with arrows, and the trypsin-specific residues (KD, boxed) (3) of An. gambiae trypsin 7 (sequence b) are boxed.
Other Differentially Expressed cDNAs.
Two of the candidate sex-specific components were confirmed by Northern blot analysis. One of these (S1) was exclusively expressed in adult males and the other (S2) in adult females. No S2 homologues were identified by data base analysis. Furthermore, 16 of the sequences obtained from several differential display experiments were subjected to developmental analysis with respect to temporal variation in gene expression during the life cycle (Fig. 2D). All but one showed significant temporal variation, ranging from purely adult to purely larval (summarized in Table 1). For some of them expression appears to be monotonic (e.g., pupal and adult sequence G3), while others show recurrent peaks (e.g., G18, which is strongly expressed in embryos and early larvae, in late larvae to early pupae and in adults).
Housekeeping and Constitutively Expressed Sequences.
Two of the studied sequences correspond to known housekeeping genes. G7 shows a clear match with the S11 protein of the 40S ribosomal subunit of yeast, plants and humans, while G5 is a homologue of the L10 protein of yeast and an insect, the midge Chironomus tentans. S4 is 100% identical to a segment of the previously described mitochondrial genome of An. gambiae (16). Finally A72, which shows no matches in the databases, is expressed constitutively during development.
DISCUSSION
The Differential Display Method.
In the short time since its introduction, differential display has proved a powerful method for identifying genes that are expressed differentially, in space or time during development. Our particular implementation is convenient and efficient and has served well for the identification of multiple components of interest, from fairly limited amounts of tissue. Resolution of differential display products on ethidium bromide-stained agarose gels significantly reduces time and effort, making it possible to perform the experiment from tissue isolation to the cloning of PCR products in less than 72 h, without the use of radioisotopes. cDNAs identified by differential display are frequently false positives and must be verified independently. Sequencing of at least the ends of the clones at an early stage is highly recommended because this permits multiple and diverse confirmatory assays based on RT–PCR. In this manner, sometimes unexpected properties can be established. For example, A16 was initially isolated as a candidate clone induced by bacterial challenge, but this was not confirmed by subsequent RT–PCR analysis; instead, whole animal developmental analysis showed that A16 was strongly expressed in late larval life. Furthermore, tissue-specific RT–PCR analysis showed that A16 expression in the adult was gut-specific. One possible explanation for the apparent initial enrichment of A16 as a result of larval infection could be a stress-induced acceleration of development, secondarily increasing the levels of A16 transcript. In this study, early sequencing followed by RT–PCR permitted us to characterize extensively 19 clones, most of which proved interesting (Table 1): 15 are temporally regulated during development, 6 are gut-specific in the adult, 2 others are modulated by feeding, 1 is induced by infection in the larva, and 1 is sex-specific.
Expressed Sequence Tags and Their Developmental Specificities.
Published nucleic acid sequences of An. gambiae are as yet limited (see P. Topalis and C. Louis, at URL: http://konops.imbb.forth.gr/AnoDB). Thus, even the sequences that are not gut-specific and show no homology to known genes are useful in their capacity as expressed sequence tags. The interest of the expressed sequence tags is substantially enhanced by their surprisingly diverse developmental expression profiles, making them potential entry points for molecular studies of development and physiology of the malaria mosquito. Furthermore, the promoters of some of the differentially expressed genes, including those that are specific for the gut may ultimately prove useful for engineering transgenic mosquitoes of desired properties, for experimental analysis or for purposes of control.
Differentially Expressed Genes of Recognizable Sequence.
Clones that are differentially expressed in the adult gut and encode proteins belonging to known families are currently the most interesting outcome of the present study. The G3-encoded protein is clearly a member of the maltase family of glycosidases (17, 30). The available sequence represents a C-terminal domain that is highly conserved in Diptera, and contains a motif (NHD) typical of the substrate binding and catalytic sites of several carbohydrate metabolizing enzymes. Most glycosidases and glycosylases studied in insects are expressed in the gut, with exceptions such as the putative maltase of Aedes aegypti which is produced in the salivary glands (30). Two maltases have already been described in An. gambiae which are also specific for the adult midgut (17). Sugars are an important energy source for the flight muscles and provide necessary nutrients for over-wintering species. The adult specificity of G3 presumably reflects the carbohydrate-rich nature of mosquito food other than blood, while G3 repression after the blood meal reflects the shift to a protein-rich food source.
G10 encompasses the complete coding region for a protein that is extremely similar to the 14-kDa subunit of V-ATPase of other metazoans. It is expressed for much of development, and in the adult it is enriched in the midgut, where it is also repressed by the blood meal. The M. sexta homologue is known to be concentrated in the apical membrane of the midgut goblet cells, where it functions in H+ secretion leading to K+/H+ antiport exchange (25, 31). In turn this serves functions such as fluid secretion, regulation of ionic concentration, and amino acid absorption from the gut lumen through the columnar epithelial cells, among which the goblet cells are interspersed.
The G14-encoded protein is a member of the large and extremely well conserved family of low molecular mass GTP-binding proteins, which are diverse in function (27). The known Drosophila homologue, Dcdc42, is implicated in axonal outgrowth and myoblast fusion (26). The G14 sequence is predominantly expressed in mosquito embryos, larvae, and early pupae, but it is also detected in the adult midgut, where it is repressed by the blood meal.
The complete coding sequences reveal that G20 and A16 are only distantly related to each other (14% amino acid identity in 172 aligned positions with 4 gaps; data not shown) but both encode lectins belonging to different families. G20 shows substantial similarity to mammalian galactose-specific lectins throughout its length, including some of the essential amino acids of the carbohydrate recognition domain involved in galactose binding (Fig. 3). It shows an ≈25% overall similarity to mammalian lactose-binding lectins, including a part of a rat intestinal lectin (22), a part of a pig lectin believed to be involved in assembly of cell-cell adherence junctions (23), and the human Galectin-7, which is involved in cell–cell and cell–matrix interactions (24). A16 reveals localized similarity to the carbohydrate recognition domains of numerous C-type (Ca2+-dependent) lectins, most significantly to human l-selectin and the human macrophage mannose receptor, and at a lower degree to a lectin of the fly, Sarcophaga peregrina (Fig. 3). l-Selectin belongs to the family of selectin cell adhesion molecules which mediate leukocyte–endothelial cell interactions (18), while the macrophage mannose receptor is involved in the discrimination of self from nonself and is thought to play an important role in the innate immune response to pathogenic microorganisms (19). The Sarcophaga lectin is induced in response to tissue injury (20). Although a particular carbohydrate binding specificity cannot be deduced from the primary sequence, A16 shows a number of interesting similarities to C-type lectins (21) (Fig. 3). Both G20 and A16 are predominantly expressed in late larvae; the dramatically increased expression of A16 in late larval instars (Fig. 2C) suggests some role in growth or differentiation processes associated with metamorphosis. In addition, both G20 and A16 are detected in the adult, exclusively in the gut. G20 is slightly repressed by the blood meal, suggesting the possibility of a function related to the diet. Conversely, A16 is slightly induced in the gut by blood feeding. Lectins are frequently found in the insect gut, and some have been implicated as recognition and opsonization factors in invertebrate innate immunity (32). However, we did not detect reproducible induction of G20 or A16 upon bacterial challenge of larvae (data not shown).
The G13 sequence is of special interest, as it clearly is a serine protease, largely associated with the adult gut and also strongly induced by bacterial challenge. It contains the highly conserved residues of the serine protease catalytic triad but lacks the trypsin specific lysine-aspartic acid pair in the carboxyl-terminal region (3). The short activation peptide (3) and significant levels of amino acid identity brings G13 closer to the mammalian serine protease families known to be involved in immune response such as kallikreins (28, 29), mast cell proteases (33), and plasminogens (34), rather than to the previously reported An. gambiae gut-specific trypsins and chymotrypsins (3). G13 shares the highest degree of similarity to an Ae. aegypti chymotrypsin of unknown function (accession no. U56423U56423). Serine proteases are known to participate in immune signaling pathways (35) as well as in the activation of immune reactive components such as prophenol oxidase to phenol oxidase during melanotic encapsulation (36). The immune responsive induction of G13 RNA levels in larvae suggest it may play a role in the mosquito immune system.
In conclusion differential display, coupled with carefully chosen and sensitive confirmatory assays for regulated expression of the isolated clones, is a powerful approach for gaining molecular tools to analyze complex biological systems. The six sequences that were shown to be associated with the adult gut will be valuable for studying the organ that represents the first major obstacle which the malaria parasite must breach to succeed in multiplying inside the mosquito vector. One of these newly identified sequences may prove to be a previously unrecognized component of the insect innate immune response.
Acknowledgments
We thank Dr. Alan Ezekowitz and Dr. Neil S. Harris for help in analyzing the sequence of A16, and Dr. Hans-Michael Müller for analyzing the sequence of G13. This work was supported by grants from the John D. and Catherine T. McArthur Foundation, the United Nations Development Programme/World Bank/World Health Organisation Special Programme for Research and Training in Tropical Diseases, the Human Frontiers Science Program, and The Hellenic Foundation of Research and Technology. G.D. and A.d.T. are recipients of fellowships from the European Union (Training and Mobility of Researchers, and Human Capital and Mobility, respectively).
Footnotes
Abbreviation: RT–PCR, reverse transcription–PCR.
Data deposition: The sequences reported in this paper have been deposited in the GenBank data base (accession numbers are given in Table 1).
References
- 1.Warburg A, Miller L H. Parasitol Today. 1991;7:179–181. doi: 10.1016/0169-4758(91)90127-a. [DOI] [PubMed] [Google Scholar]
- 2.Collins F H, Sakai R K, Vernick K D, Paskewitz S, Seeley D C, Miller L H, Collins W E, Campbell C C, Gwadz R W. Science. 1986;234:607–610. doi: 10.1126/science.3532325. [DOI] [PubMed] [Google Scholar]
- 3.Müller H-M, Catteruccia F, Vizioli J, della Torre A, Crisanti A. Exp Parasitol. 1996;81:371–385. doi: 10.1006/expr.1995.1128. [DOI] [PubMed] [Google Scholar]
- 4.Collins F H. Parasitol Today. 1994;10:370–376. doi: 10.1016/0169-4758(94)90221-6. [DOI] [PubMed] [Google Scholar]
- 5.Williams J G K, Hanafey M K, Rafalski J A, Tingey S V. Methods Enzymol. 1993;218:704–740. doi: 10.1016/0076-6879(93)18053-f. [DOI] [PubMed] [Google Scholar]
- 6.Welsh J, Chada K, Dalal S S, Cheng R, Ralph D, McClelland M. Nucleic Acids Res. 1992;20:4965–4970. doi: 10.1093/nar/20.19.4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang P, Pardee A B. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- 8.Richman A M, Bulet C, Hetru C, Barillas-Mury C, Hoffman J A, Kafatos F C. Insect Mol Biol. 1996;5:203–210. doi: 10.1111/j.1365-2583.1996.tb00055.x. [DOI] [PubMed] [Google Scholar]
- 9.Devereux J, Haeberli P, Smithies O. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 11.Church G M, Gilbert W. Proc Natl Acad Sci USA. 1984;81:1991–1995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.della Torre A, Favia G, Mariotti G, Coluzzi M, Mathiopoulos K D. Genetics. 1996;143:1307–1311. doi: 10.1093/genetics/143.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng L, Saunders R D C, Fortini D, della Torre A, Coluzzi M, Glover D M, Kafatos F C. Proc Natl Acad Sci USA. 1991;88:11187–11191. doi: 10.1073/pnas.88.24.11187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salazar C E, Hamm D M, Kumar V, Collins F H. Nucleic Acids Res. 1993;21:4147. doi: 10.1093/nar/21.17.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salazar C E, Hamm D M, Wesson D M, Beard C B, Kumar V, Collins F H. Insect Mol Biol. 1994;3:1–13. doi: 10.1111/j.1365-2583.1994.tb00145.x. [DOI] [PubMed] [Google Scholar]
- 16.Beard C B, Hamm D M, Collins F H. Insect Mol Biol. 1993;2:103–124. doi: 10.1111/j.1365-2583.1993.tb00131.x. [DOI] [PubMed] [Google Scholar]
- 17.Zheng L, Whang L H S, Kumar V, Kafatos F C. Exp Parasitol. 1996;81:272–283. doi: 10.1006/expr.1995.1118. [DOI] [PubMed] [Google Scholar]
- 18.Siegelman H M, Weissman I L. Proc Natl Acad Sci USA. 1989;86:5562–5566. doi: 10.1073/pnas.86.14.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taylor M E, Conary J L, Lennartz M R, Stahl P D, Drickamer K. J Biol Chem. 1990;265:12156–12162. [PubMed] [Google Scholar]
- 20.Takahashi H, Komano H, Kawaguchi N, Kitamura N, Nakanishi S, Natori S. J Biol Chem. 1985;260:12228–12233. [PubMed] [Google Scholar]
- 21.Drickamer K. Curr Opin Struct Biol. 1993;3:393–400. [Google Scholar]
- 22.Oda Y, Herrmann J, Gitt M A, Turck C W, Burlingame A L, Barondes S H, Seffler H. J Biol Chem. 1993;268:5929–5939. [PubMed] [Google Scholar]
- 23.Chiu M L, Parry D A D, Feldman S R, Klapper D G, O’Keefe E J. J Biol Chem. 1994;269:31770–31776. [PubMed] [Google Scholar]
- 24.Madsen P, Rasmussen H H, Flint T, Gromov P, Kruse T A, Honore B, Vorum H, Celis J E. J Biol Chem. 1995;270:5823–5829. doi: 10.1074/jbc.270.11.5823. [DOI] [PubMed] [Google Scholar]
- 25.Jäger D, Novak F J S, Harvey W R, Wieczorek H, Klein U. J Exp Biol. 1996;199:1019–1027. doi: 10.1242/jeb.199.5.1019. [DOI] [PubMed] [Google Scholar]
- 26.Luo L, Liao J, Jan L Y, Jan Y N. Genes Dev. 1994;8:1787–1802. doi: 10.1101/gad.8.15.1787. [DOI] [PubMed] [Google Scholar]
- 27.Shinjo K, Koland J G, Hart M J, Narasimhan V, Johnson D I, Evans T, Cerione R A. Proc Natl Acad Sci USA. 1990;87:9853–9857. doi: 10.1073/pnas.87.24.9853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohen W M, Wu H F, Featherstone G L, Jenzano J W, Lundblad R L. Biochem Biophys Res Commun. 1991;176:315–320. doi: 10.1016/0006-291x(91)90926-x. [DOI] [PubMed] [Google Scholar]
- 29.Mason A J, Evans B A, Cox D R, Shine J, Richards R I. Nature (London) 1983;303:300–307. doi: 10.1038/303300a0. [DOI] [PubMed] [Google Scholar]
- 30.James A A, Blackmer K, Racioppi J V. Gene. 1989;75:73–83. doi: 10.1016/0378-1119(89)90384-3. [DOI] [PubMed] [Google Scholar]
- 31.Graf R, Lepier A, Harvey W R, Wieczorek H. J Biol Chem. 1994;268:3767–3774. [PubMed] [Google Scholar]
- 32.Ham P J. In: Advances in Disease Vector Research. Harris K, editor. Vol. 9. New York: Springer; 1992. pp. 101–149. [Google Scholar]
- 33.McNeil H P, Reynolds D S, Schiller V, Ghildyal N, Gurley D S, Austen K F, Stevens R L. Proc Natl Acad Sci USA. 1992;89:11174–11178. doi: 10.1073/pnas.89.23.11174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamilton J A, Whitty G A, Wojta J, Gallichio M, McGrath K, Ianches G. Cell Immunol. 1993;152:7–17. doi: 10.1006/cimm.1993.1263. [DOI] [PubMed] [Google Scholar]
- 35.Hoffmann J A. Curr Opin Immunol. 1995;7:4–10. doi: 10.1016/0952-7915(95)80022-0. [DOI] [PubMed] [Google Scholar]
- 36.Hultmark D. Trends Genet. 1993;9:178–183. doi: 10.1016/0168-9525(93)90165-e. [DOI] [PubMed] [Google Scholar]