

Abstract
We have analyzed the expression of the breast cancer susceptibility gene, Brca2, in mammary epithelial cells as a function of proliferation and differentiation. Our results demonstrate that Brca2 mRNA expression is tightly regulated during mammary epithelial proliferation and differentiation, and that this regulation occurs coordinately with Brca1. Specifically, Brca2 mRNA expression is up-regulated in rapidly proliferating cells; is down-regulated in response to serum deprivation; is expressed in a cell cycle-dependent manner, peaking at the G1/S boundary; and is up-regulated in differentiating mammary epithelial cells in response to glucocorticoids. In each case, an identical pattern of expression was observed for Brca1. These results indicate that proliferative stimuli modulate the mRNA expression of these two breast cancer susceptibility genes. In addition, the coordinate regulation of Brca1 and Brca2 revealed by these experiments suggests that these genes are induced by, and may function in, overlapping regulatory pathways involved in the control of cell proliferation and differentiation.
Keywords: Brca2 expression, Brca1 expression, mammary gland
The inheritance of germ-line mutations in autosomal dominant susceptibility genes appears to be responsible for 5–10% of all breast cancer cases (1, 2). The breast cancer susceptibility gene, BRCA2, was recently isolated by positional cloning and is predicted to encode a 3418-aa polypeptide that lacks significant homology to previously described proteins (3–5). Like the breast and ovarian cancer susceptibility gene, BRCA1, germ-line mutations in BRCA2 are believed to account for up to half of families with inherited breast cancer. In contrast to BRCA1, however, germ-line mutations in BRCA2 predispose male carriers to an increased risk of breast cancer, and female carriers to an increase in ovarian cancer risk which is less pronounced than that associated with germ-line BRCA1 mutations (3, 6). In addition, a variety of other cancers appear to occur at higher frequencies in BRCA2 families, including carcinomas of the pancreas, prostate, and colon (7–10). Because most breast tumors arising in patients with germ-line BRCA2 mutations have been found to exhibit loss of the wild-type BRCA2 allele, this gene is believed to function as a tumor suppressor (9, 11). Nevertheless, little is currently known about the function or regulation of this gene.
The dramatically elevated risks of breast cancer observed in women carrying germ-line mutations in either of the familial breast cancer susceptibility genes, BRCA1 or BRCA2, suggest that these genes play important roles in the regulation of mammary epithelial cell growth. This suggestion is consistent with the observation that the murine homologue of BRCA1 is widely expressed in proliferating and differentiating cell types in the mouse during embryonic and mammary gland development, and in adult tissues (12, 13). This hypothesis is further supported by the findings that homozygous mutations in Brca1 result in early embryonic lethality in mice and that overexpression of BRCA1 appears to inhibit the growth of breast and ovarian cancer cell lines in vivo and in vitro (14, 15). We have previously shown that Brca1 mRNA expression is up-regulated in the mammary epithelium during developmental stages characterized by rapid cellular proliferation and differentiation, namely puberty and pregnancy (12). Recently, we have shown that the spatial and temporal pattern of Brca2 mRNA expression in the mouse is strikingly similar to that exhibited by Brca1 during embryonic development and in adult tissues (unpublished data). Specifically, Brca2 is expressed at high levels in rapidly proliferating and differentiating cellular compartments, including those in the breast, such as terminal end buds during puberty and differentiating alveoli during pregnancy. Nevertheless, despite the intriguing similarities in the developmental regulation of Brca1 and Brca2 expression in the mammary gland, the basis for this similarity is currently unknown, as is the extent to which these cancer susceptibility genes may be regulated by or involved in the processes of proliferation and differentiation in nontransformed mammary epithelial cells.
MATERIALS AND METHODS
Cell Culture.
The nontransformed mammary epithelial cell line, NMuMG, was obtained from ATCC. Additional cell lines were derived from mammary gland tumors or hyperplastic lesions that arose in transgenic mice carrying the MMTV-c-myc transgene, the MMTV-int-2 transgene, or the MMTV-neu/NT transgene as described (16). Cells were grown in DMEM supplemented with 10% bovine calf serum, 2 mM lglutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin.
HC11 cells were the gift of J. Rosen (Baylor College of Medicine, Houston) and B. Groner (Institute for Experimental Cancer Research, Freiburg, Germany) and were grown in RPMI 1640 medium containing 10% bovine calf serum, 5 μg/ml insulin (Sigma), 10 ng/ml epidermal growth factor (Sigma), 2 mM l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. HC11 cells were induced to differentiate 3 days after reaching confluence by adding differentiation medium containing 10% bovine calf serum, 5 μg/ml insulin, 1 μg/ml hydrocortisone (or 1 μM dexamethasone), and 5 μg/ml ovine prolactin (National Hormone and Pituitary Program). Differentiating cells were refed daily.
Cell proliferation was quantitated by tritiated thymidine incorporation and was performed essentially as described (17). Tritiated thymidine and cell counting assays were performed in triplicate. Cell cycle synchronization with nocodazole, mimosine, and hydroxyurea was performed as described (18).
RNA Analysis.
Northern blot hybridization and RNase protection analysis were performed as described (12). DNA fragments used as probes for Northern blot hybridization included a 1.0-kb region of mouse Brca1 cDNA corresponding to amino acids 760-1099 of human exon 11, a 551-bp region of mouse Brca2 cDNA corresponding to nucleotides 281–853 of the human BRCA2 cDNA, nucleotides 181–719 of mouse β-casein, or chicken histone H2B (Oncor). Riboprobes for RNase protection analysis included an approximately 230-bp PCR fragment of the mouse Brca1 cDNA as described (12) or an approximately 240-bp fragment of mouse Brca2 corresponding to amino acids 127–208 of human BRCA2 subcloned into the TA cloning vector (Invitrogen). Brca2 reactions were processed as described for Brca1, except that samples were hybridized at 52°C.
RESULTS
In vivo studies in the mouse have indicated that both Brca1 and Brca2 are expressed in rapidly proliferating and differentiating cell types, including those found in the breast during puberty and early pregnancy (ref. 12 and unpublished data). In addition, the observation that inherited mutations in either BRCA1 or BRCA2 predispose women to adenocarcinoma of the breast suggests that these genes may function in similar or overlapping regulatory pathways in mammary epithelial cells. To begin to analyze the regulation of Brca2 expression in mammary epithelial cells, to examine the mechanisms underlying the similarity in Brca1 and Brca2 expression patterns during mammary gland development, and to test the hypothesis that both Brca1 and Brca2 expression are regulated during the processes of proliferation and differentiation, we have characterized the expression of these putative tumor suppressor genes as a function of proliferation and differentiation in mammary epithelial cells in vitro. cDNA clones encoding the amino terminus of the murine homologue of BRCA2 were isolated and characterized (unpublished data). The murine cDNA sequence in this region shows significant homology only to human BRCA2 and is 78% identical to human BRCA2 at the nucleotide level and 80% similar and 70% identical at the amino acid level. Northern hybridization analysis of murine tissues using this cDNA as probe detected a single mRNA with length and tissue distribution similar to those detected by human BRCA2 (unpublished data). Taken together, the similarities in sequence homology, pattern of Northern hybridization, mRNA size, and tissue distribution of expression strongly suggest that this murine cDNA sequence is derived from the bona fide murine homologue of BRCA2.
Brca2 mRNA levels were analyzed in NIH 3T3 fibroblasts and in a panel of mammary epithelial cell lines that were either actively proliferating or confluent (Fig. 1A). Mammary epithelial cell lines tested included nontransformed cell lines as well as transformed lines derived from breast tumors arising in transgenic mice overexpressing activated c-neu, c-myc, or int-2 (16). RNase protection analysis revealed that in each case Brca2 mRNA was highly expressed in proliferating cells and markedly down-regulated in confluent cells. Interestingly, this same pattern of expression was observed for Brca1. Histone H2B mRNA levels were also down-regulated in confluent cells, as expected from its S-phase-dependent pattern of expression (Fig. 1A and ref. 19). The down-regulation of Brca2 mRNA levels in confluent cells occurred in nontransformed and transformed cells, and in transgenic breast cancer cell lines irrespective of the particular oncogene present. Because multiple phenomena could be responsible for the observed reduction in Brca2 expression in confluent cells, including the effects of contact inhibition and growth factor depletion, Brca2 expression was analyzed in nontransformed mammary epithelial cell lines subjected to serum starvation (Fig. 1B). As expected, serum starvation resulted in a marked decrease in proliferation, as reflected by decreases in histone H2B expression and [3H]thymidine incorporation (Fig. 1B and data not shown). Similarly, Brca2 expression was markedly down-regulated in response to serum starvation in each of these cell lines (Fig. 1B). Again, the same pattern of expression was observed for Brca1.
Figure 1.
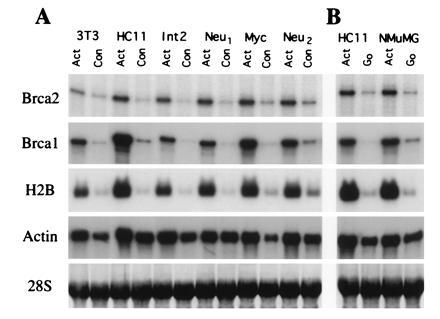
Brca1 and Brca2 expression are a function of proliferative state. (A) mRNA expression of Brca1, Brca2, histone H2B, and actin in actively growing (Act) or confluent (Con) murine cell lines. Cell line RNAs analyzed were prepared from NIH 3T3 fibroblasts (3T3), nontransformed mammary epithelial cells (HC11), or transformed mammary epithelial cells derived from breast tumors arising in transgenic mice overexpressing either activated c-neu, c-myc, or int-2, as indicated. (B) mRNA expression of Brca1, Brca2, histone H2B, and actin in actively growing (Act) or serum starved (G0) nontransformed mammary epithelial cell lines, as indicated. mRNA expression was quantitated by RNase protection (Brca1, Brca2, and actin) or Northern blot hybridization (histone H2B). The 28S RNA band from an ethidium bromide-stained nitrocellulose blot used as a loading control is shown.
To further explore the relationship between changes in proliferation and the regulation of Brca2 expression, the nontransformed mammary epithelial cell line, HC11, was synchronized in G0 by serum starvation. Serum-starved cells were stimulated to re-enter the cell cycle by the addition of serum. [3H]Thymidine incorporation and steady-state levels of Brca2, Brca1, histone H2B, and β-actin mRNA were analyzed as a function of time after serum addition (Fig. 2 A and B). HC11 cells entered S phase within 24 hr after their release by serum re-addition as determined by [3H]thymidine incorporation (Fig. 2B). Consistent with this, maximal histone H2B mRNA expression (which occurs immediately before entrance into S phase) occurred 15 hr after serum re-addition (Fig. 2A). Steady-state levels of Brca2 mRNA began to rise 9 hr after serum re-addition, peaked at 12 hr, and remained at maximal levels 15 hr after serum re-addition, just before the G1/S transition. Again, Brca1 expression demonstrated a temporal pattern remarkably similar to that exhibited by Brca2, rising at 9 hr and peaking 15 hr after serum re-addition. Essentially identical results were obtained after synchronization of the nontransformed mammary epithelial cell line, NMuMG (Fig. 2 C and D). After serum re-addition, p53 mRNA levels peaked in NMuMG cells just before peak expression of Brca2 and Brca1, consistent with previous descriptions of p53 expression during passage through G1 (20).
Figure 2.
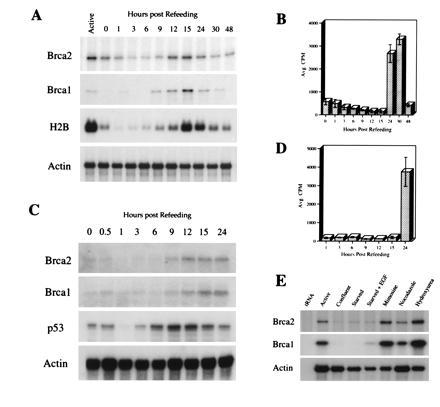
Brca1 and Brca2 mRNA expression are cell cycle-dependent. (A) mRNA expression levels of Brca1, Brca2, histone H2B, and actin in actively growing (Active) HC11 cells or in serum-starved cells at the times indicated after refeeding with 10% serum. mRNA expression was quantitated by RNase protection (Brca1, Brca2, and actin) or Northern blot hybridization (histone H2B). (B) [3H]Thymidine incorporation in serum-starved HC11 cells from A at the times indicated after refeeding with 10% serum. (C) mRNA expression of Brca1, Brca2, p53, and actin in actively growing (Active) NMuMG cells or in serum-starved cells at the times indicated after refeeding with 10% serum. mRNA expression was quantitated by RNase protection (p53 and actin) or Northern hybridization (Brca1 and Brca2). (D) [3H]Thymdine incorporation in serum-starved NMuMG cells from C at the times indicated after refeeding with 10% serum. (E) mRNA expression of Brca1, Brca2, and actin in HC11 cells that were either actively growing (Active), confluent, serum-starved, serum-starved and treated with epidermal growth factor, or synchronized by treatment with mimosine, nocodazole, or hydroxyurea. mRNA expression was quantitated by RNase protection.
To distinguish bona fide cell cycle regulation from mitogenic effects intrinsic to serum starvation/refeeding protocols, HC11 cells were cultured in the presence of mimosine, nocodazole, or hydroxyurea to determine the level of Brca2 mRNA expression in cells arrested at specific points in the cell cycle (Fig. 2E). As before, steady-state levels of Brca2 mRNA were down-regulated in confluent cells and in actively growing cells subjected to serum starvation, irrespective of the presence of epidermal growth factor. Steady-state levels of Brca2 mRNA in HC11 cells arrested late in G1 after treatment with mimosine, or early in S phase after treatment with hydroxyurea, were higher than those observed in actively proliferating cells or in cells arrested at metaphase by treatment with nocodazole. Again, a similar pattern of expression was observed for Brca1. These observations are consistent with those made above in serum-starved HC11 cells permitted to progress through the cell cycle in a synchronous fashion after serum refeeding. Taken together, these findings are compatible with a model in which proliferative stimuli modulate both Brca1 and Brca2 mRNA expression in a cell cycle-dependent manner.
The coexpression of Brca1 and Brca2 in mammary epithelial cells described above, coupled with the coordinate developmental regulation of Brca1 and Brca2 mRNA expression in the mammary gland, suggested to us that the expression of these molecules may be modulated during mammary epithelial differentiation as well as proliferation (refs. 12 and 13 and unpublished data). To test this hypothesis, we analyzed Brca2 expression, as well as Brca1 expression, in vitro in postconfluent HC11 mammary epithelial cells induced to differentiate in response to the lactogenic hormones prolactin, insulin, and hydrocortisone (Fig. 3 A and B). As above, the conversion of actively growing cells to the postconfluent state was accompanied by the down-regulation of Brca1 and Brca2 mRNA expression as well as by concomitant decreases in proliferation as reflected by [3H]thymidine incorporation and steady-state levels of histone H2B mRNA (Fig. 3 A and B). Proliferation remained low in differentiating cells relative to actively growing cells throughout the differentiation time course, as expected from their postconfluent state. In light of our previous observations, these findings predicted that expression levels of both Brca2 and Brca1 would remain low in differentiating HC11 cells if their proliferative state was the primary determinant of expression. In contrast to this expectation, Brca2 expression was up-regulated after treatment of postconfluent HC11 cells with lactogenic hormones (Fig. 3A). Moreover, the temporal pattern of Brca1 mRNA induction was again remarkably similar to that exhibited by Brca2. In differentiating HC11 cells, steady-state levels of Brca2 mRNA began to rise at day 3 and peaked at day 7 of induction with lactogenic hormones. Similarly, Brca1 mRNA expression began to rise at day 4 and, like Brca2, peaked at day 7 of induction. Steady-state levels of both Brca1 and Brca2 mRNAs were as high in postconfluent, differentiating cells as in actively growing cells despite the fact that proliferation remained low and essentially unchanged during the course of differentiation. These observations strongly suggest that the up-regulation of Brca1 and Brca2 expression observed in differentiating mammary epithelial cells occurs by a proliferation-independent pathway. As such, these results further support the hypothesis that the mRNA expression of each of these genes is regulated in mammary epithelial cells as a function both of proliferation and of differentiation.
Figure 3.

Brca1 and Brca2 expression are induced by glucocorticoids during mammary epithelial differentiation. (A) mRNA expression of Brca1, Brca2, histone H2B, β-casein, and actin in HC11 mammary epithelial cells that were either actively growing (Active) or postconfluent at the times indicated after induction with lactogenic hormones (insulin, hydrocortisone, and prolactin) in the presence of whole serum. mRNA expression was quantitated by RNase protection (Brca1 and actin) or Northern blot hybridization (β-casein, Brca2, and histone H2B). The 28S RNA band from an ethidium bromide-stained nitrocellulose blot used as a loading control is shown. (B) [3H]Thymdine incorporation (corrected for number of cells) of actively growing or differentiating HC11 cells shown in A. (C) Hormonal requirements for Brca1 and Brca2 up-regulation in HC11 cells. mRNA expression of Brca1, Brca2, histone H2B, β-casein, and actin as measured by Northern blot hybridization in HC11 mammary epithelial cells that were either actively growing (Active), confluent for 3 days in growth medium (Con), or postconfluent and treated with the indicated hormones for 7 days in differentiation medium. HC, hydrocortisone; Ins, insulin; Prl, prolactin.
If Brca1 and Brca2 up-regulation is an intrinsic part of differentiation in HC11 cells, then the hormonal requirements for this up-regulation should parallel those for differentiation. Conversely, if the hormonal requirements for Brca1 and Brca2 up-regulation are a subset of those required for differentiation, this would identify hormone-induced signal transduction pathways likely to be involved in the regulated expression of these putative tumor suppressor genes. In addition, the similarities or differences in the specific hormonal requirements for the induction of each of these genes would be expected to shed light on the basis for their apparent coordinate regulation. To compare the hormonal requirements for the up-regulation of Brca2 expression in HC11 cells with those for Brca1, and to compare each of those with the hormonal requirements for differentiation, we treated HC11 cells with either complete differentiation medium containing the lactogenic hormones prolactin, hydrocortisone, and insulin, or with medium lacking one or more of these hormones (Fig. 3C). As expected, all three lactogenic hormones were required to induce the differentiation of HC11 cells as manifested by β-casein expression. Brca2 mRNA expression was induced to maximal levels in postconfluent cells after treatment with complete differentiation medium containing insulin, prolactin, and either of the glucocorticoids, hydrocortisone or dexamethasone (Figs. 3C and 4B). A similar, induction of Brca2 expression occurred in response to the combination of insulin and either hydrocortisone or dexamethasone (Figs. 3C and 4B). Again, the steady-state level of Brca2 mRNA in differentiating mammary epithelial cells was as high as that found in actively growing cells despite the fact that proliferation rates remained low, as reflected by histone H2B expression levels. In contrast, treatment of postconfluent HC11 cells with either insulin alone or with insulin plus prolactin had no effect on Brca2 mRNA levels. These results suggest that glucocorticoids are primarily responsible for the proliferation-independent up-regulation of Brca2 expression in mammary epithelial cells. Strikingly, the regulation of Brca1 expression in response to each of these hormonal combinations was essentially indistinguishable from that observed for Brca2. This observation provides additional evidence for the parallel regulation of Brca1 and Brca2 expression.
Figure 4.
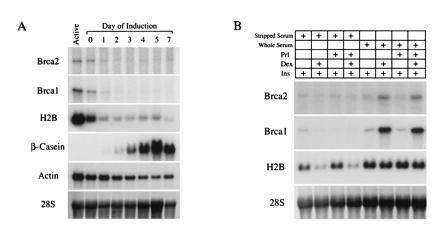
The up-regulation of Brca1 and Brca2 by glucocorticoids requires a serum factor(s). (A) mRNA expression of Brca1, Brca2, histone H2B, β-casein, and actin in HC11 mammary epithelial cells that were either actively growing (Active) or postconfluent at the times indicated after induction with lactogenic hormones (insulin, hydrocortisone, and prolactin) in the presence of charcoal-stripped serum. The 28S RNA band is shown from an ethidium bromide-stained nitrocellulose blot as a loading control. (B) Hormonal requirements for Brca1 and Brca2 up-regulation in HC11 cells in charcoal-stripped or whole serum. mRNA expression of Brca1, Brca2, and histone H2B in postconfluent HC11 cells that were treated in the presence of charcoal-stripped or whole serum with the hormones indicated for 7 days in differentiation medium. Dex, dexamethasone; Ins, insulin; Prl, prolactin.
Treatment of postconfluent HC11 cells with lactogenic hormones in the presence of charcoal-stripped serum, rather than whole serum, also resulted in the efficient differentiation of HC11 cells as manifested by the accumulation of β-casein mRNA (Fig. 4A). In contrast to results obtained in whole serum, however, neither Brca2 nor Brca1 mRNA levels were up-regulated under these conditions, but rather paralleled decreases in proliferation as reflected by histone H2B expression. This was confirmed by treating postconfluent HC11 cells with either complete differentiation medium or medium lacking one or more hormones in the presence of either stripped or whole serum (Fig. 4B). As before, treatment of postconfluent HC11 cells with glucocorticoids in the presence of whole serum resulted in the up-regulation of both Brca1 and Brca2 expression in the absence of changes in proliferation. However, no significant changes in the expression of either Brca1 or Brca2 was observed after treatment of postconfluent HC11 cells with glucocorticoids in the presence of stripped serum. This suggests that a factor(s) that is present in serum and removed by charcoal stripping is required for the proliferation-independent up-regulation of Brca2, as well as Brca1, expression in differentiating mammary epithelial cells. Moreover, these results provide additional evidence for the coordinate regulation of Brca1 and Brca2. Finally, although these results indicate that the induction of Brca1 and Brca2 expression is not required for HC11 mammary epithelial differentiation, the up-regulation of these genes may nevertheless occur during differentiation in vivo, because whole serum is likely to represent a closer approximation of the in vivo environment of the mammary epithelium. This hypothesis is supported by in vivo observations of Brca1 and Brca2 expression during mammary gland development (refs. 12 and 13 and unpublished data).
DISCUSSION
We have analyzed the expression of Brca1 and Brca2 in mammary epithelial cells as a function of proliferation and differentiation to test our hypothesis that the expression of these two breast cancer susceptibility genes is regulated during these processes. Using in vitro systems in which immortalized cell lines can be induced to proliferate or differentiate, we found that Brca1 and Brca2 mRNAs are expressed at high levels in rapidly proliferating cells, are down-regulated in response to growth factor deprivation, are induced in a cell cycle-dependent manner at the G1/S boundary, and are specifically up-regulated in differentiating mammary epithelial cells in response to glucocorticoids. These results strongly suggest that Brca1 and Brca2 expression are coordinately regulated, that expression of Brca1 and Brca2 is regulated in mammary epithelial cells as a function of both proliferation and differentiation, and that these modes of regulation may occur independently.
The parallel regulation of Brca1 and Brca2 expression observed during mammary epithelial proliferation and differentiation suggests that the mRNA expression of these genes is controlled by similar agents or pathways. In light of evidence that BRCA1 may regulate proliferation, these similarities further raise the possibility that Brca1 and Brca2 function in the same or overlapping regulatory pathways involved in the control of cell proliferation and differentiation. This is consistent with the fact that mammary epithelial cells are affected similarly by inherited mutations in either BRCA1 or BRCA2. In addition, the nearly identical timing with which the induction of Brca1 and Brca2 expression occurs during the cell cycle after serum stimulation of synchronized cells, and during mammary epithelial differentiation, raises the possibility that the expression of these genes is induced by a common factor(s). Although there are no extended regions of nucleotide or amino acid homology between BRCA1 and BRCA2, each is predicted to be a large protein, each cDNA contains a single unusually large exon, and each coding region is unusually A+T-rich (4, 21). Nevertheless, despite the marked similarities in the patterns of regulation of these two genes, the fact that germ-line mutations in BRCA1 predispose carriers to increased risks of breast cancer despite the presence of wild-type copies of BRCA2, and vice versa, suggests that the functions of these two genes are not entirely redundant. This hypothesis is supported by the fact that germ-line mutations in BRCA1 and BRCA2 have distinct phenotypes with respect to the incidence of ovarian cancer, male breast cancer, and pancreatic cancer.
It has previously been shown that antisense-mediated reduction in BRCA1 expression in human mammary epithelial cells results in their increased proliferation (22, 23). In addition, overexpression of BRCA1 appears to inhibit the growth of breast and ovarian cancer cell lines in vivo and in vitro (14). Each of these findings suggests that BRCA1 negatively regulates proliferation in these cell types, and is consistent both with the occurrence of LOH in familial breast cancers in BRCA1 carriers and with the putative role of BRCA1 as a tumor suppressor gene. In this regard, the observations that steady-state Brca1 and Brca2 mRNA levels are up-regulated in rapidly growing cells, are down-regulated in confluent and serum-starved cells, and are expressed at highest levels in synchronized cells at the G1/S transition indicate that the mRNA expression of these putative tumor suppressor genes responds to changes in growth conditions. Moreover, these observations further raise the possibility that Brca1 and Brca2 may be involved in regulating progression through the cell cycle and/or entrance into S phase after growth factor stimulation.
The observation that the induction of Brca1 and Brca2 expression in differentiating mammary epithelial cells occurs in the absence of changes in proliferation demonstrates that the up-regulation of each of these genes in mammary epithelial cells may occur by proliferation-independent as well as proliferation-dependent pathways. If the regulation of Brca1 and/or Brca2 expression during mammary epithelial cell proliferation and differentiation reflects these genes’ functional involvement in these processes, our findings provide additional support to the hypothesis that each of these genes may play a role in mammary epithelial differentiation that is distinct from its role in proliferation. Interestingly, comparison of Brca1 and Brca2 regulation in mammary epithelial cells with that in other differentiating cell types, such as preadipocytes, reveals that although Brca1 and Brca2 are coordinately regulated as a function of proliferation in preadipocytes, these genes are not coordinately regulated during adipocyte differentiation (data not shown). This suggests that the coordinate up-regulation of Brca1 and Brca2 during differentiation may be specific for certain cell types, including mammary epithelial cells.
We have previously shown that both Brca1 and Brca2 expression are induced in the mammary gland during puberty and pregnancy, periods of development that are associated with increases in epithelial proliferation (ref. 12 and unpublished data). The data presented here confirm that Brca1 and Brca2 mRNA expression are induced in rapidly proliferating cells and are down-regulated in quiescent cells. The induction of Brca1 and Brca2 expression in actively growing cells, coupled with BRCA1’s potential function as a negative regulator of proliferation, suggests the possible existence of a regulatory loop in which proliferation induces BRCA1 and BRCA2 expression which, in turn, negatively regulate proliferation. The existence of such a homeostatic loop would imply that the proliferation-induced up-regulation of Brca1 and Brca2 expression constitutes a protective response tending to decrease breast cancer risk. This is consistent with the fact that these genes are induced in the mammary gland during developmental stages (puberty and pregnancy) that are associated with both increased cellular proliferation and increased breast cancer risk (24, 25). Such a model would predict that women exhibiting lower levels of Brca1 or Brca2 activity may have an even further increased risk of breast cancer associated with these developmental states. Interestingly, recent epidemiological studies suggest that women with a positive family history of breast cancer may experience a significantly greater increase in breast cancer risk associated with their first pregnancy than women without a family history of breast cancer (26). Whether this phenomenon results from abrogating a protective effect normally exerted by Brca1 and/or Brca2 during pregnancy awaits further study.
Acknowledgments
We thank Nate Chodosh for primary fibroblasts, Mitch Lazar for insightful discussions, Jeffrey Rosen and Bernd Groner for HC11 cells, Philip Leder for transgenic cell lines, and members of the Chodosh laboratory. Prolactin (AFP-10677C) was obtained through the National Hormone and Pituitary Program. Sequence analysis was performed at the National Center for Biotechnology Information using the BLAST network service. This work was supported in part by the Charles E. Culpeper Foundation. L.A.C. is a Charles E. Culpeper Medical Scholar.
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank data base (accession no. 472947).
References
- 1.Newman B, Austin M A, Lee M, King M-C. Proc Natl Acad Sci USA. 1988;85:1–5. doi: 10.1073/pnas.85.9.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Claus E B, Risch N, Thompson W D. Am J Hum Genet. 1991;48:232–241. [PMC free article] [PubMed] [Google Scholar]
- 3.Wooster R, Neuhausen S L, Mangion J, Quirk Y, Ford D, et al. Science. 1994;265:2088–2090. doi: 10.1126/science.8091231. [DOI] [PubMed] [Google Scholar]
- 4.Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J. Nature (London) 1995;378:789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 5.Tavtigian S V, Simard J, Rommens J, Couch F, Shattuck-Eidens D, et al. Nat Genet. 1996;12:333–337. doi: 10.1038/ng0396-333. [DOI] [PubMed] [Google Scholar]
- 6.Thorlacius S, Tryggvodorttir L, Olafsdottir G H, Jonasson J G, Ogmundsdottir H M, Tulinius H. Lancet. 1995;346:544–545. doi: 10.1016/s0140-6736(95)91383-1. [DOI] [PubMed] [Google Scholar]
- 7.Thorlacius S, Olafsdottir G, Tryggvadottir L, Neuhausen S, Jonasson J G, Tavtigian S V, Tulinius H, Ogmundsdottir H M, Eyfjord J E. Nat Genet. 1996;13:117–119. doi: 10.1038/ng0596-117. [DOI] [PubMed] [Google Scholar]
- 8.Phelan C M, Lancaster J M, Tonin P, Grumbs C, Cochran C, et al. Nat Genet. 1996;13:120–122. doi: 10.1038/ng0596-120. [DOI] [PubMed] [Google Scholar]
- 9.Gudmundsson J, Johannesdottir G, Bergthorsson J T, Arason A, Ingvarsson S, Egilsson V. Cancer Res. 1995;55:4830–4832. [PubMed] [Google Scholar]
- 10.Tonin P, Ghadirian P, Phelan C, Lenoir G M, Lynch H T, Letendre F. J Med Genet. 1995;32:982–984. doi: 10.1136/jmg.32.12.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins N, McManus R, Wooster R, Mangion J, Seal S, Lakhani S R. Oncogene. 1995;10:1673–1675. [PubMed] [Google Scholar]
- 12.Marquis S T, Rajan J V, Wynshaw-Boris A, Xu J, Yin G-Y, Abel K J, Weber B L, Chodosh L A. Nat Genet. 1995;11:17–26. doi: 10.1038/ng0995-17. [DOI] [PubMed] [Google Scholar]
- 13.Lane T F, Deng C X, Elson A, Lyu M S, Kozak C A, Leder P. Genes Dev. 1995;9:2712–2722. doi: 10.1101/gad.9.21.2712. [DOI] [PubMed] [Google Scholar]
- 14.Holt J, Thompson M, Szabo C, Robinson-Benion C, Arteaga C, King M-C, Jensen R. Nat Genet. 1996;12:298–302. doi: 10.1038/ng0396-298. [DOI] [PubMed] [Google Scholar]
- 15.Gowen L C, Johnson B L, Latour A M, Sulik K K, Koller B. Nat Genet. 1996;12:191–194. doi: 10.1038/ng0296-191. [DOI] [PubMed] [Google Scholar]
- 16.Morrison B W, Leder P. Oncogene. 1994;9:3417–3426. [PubMed] [Google Scholar]
- 17.Freshney R I. Culture of Animal Cells. New York: Wiley–Liss; 1994. pp. 277–278. [Google Scholar]
- 18.Krek W, DeCaprio J. Methods Enzymol. 1995;254:114–124. doi: 10.1016/0076-6879(95)54009-1. [DOI] [PubMed] [Google Scholar]
- 19.Stein G, Stein J, van Wijnen A, Lian J. J Cell Biochem. 1994;54:393–404. doi: 10.1002/jcb.240540406. [DOI] [PubMed] [Google Scholar]
- 20.Reich N C, Levine A J. Nature (London) 1984;308:199–201. doi: 10.1038/308199a0. [DOI] [PubMed] [Google Scholar]
- 21.Miki Y, Swensen J, Shattuck-Eidens D, Futreal P A, Harshman K, et al. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 22.Thompson M E, Jensen R A, Obermiller P S, Page D L, Holt J T. Nat Genet. 1995;9:444–450. doi: 10.1038/ng0495-444. [DOI] [PubMed] [Google Scholar]
- 23.Rao V N, Shao N S, Ahmad M, Reddy R S P. Oncogene. 1996;12:523–528. [PubMed] [Google Scholar]
- 24.Boice J D, Jr, Monson R R. J Natl Cancer Inst. 1977;59:823–832. doi: 10.1093/jnci/59.3.823. [DOI] [PubMed] [Google Scholar]
- 25.Lambe M, Hsieh C-C, Tricholpoulos D, Ekbom A, Pavia M, Adami H-O. N Engl J Med. 1994;331:5–9. doi: 10.1056/NEJM199407073310102. [DOI] [PubMed] [Google Scholar]
- 26.Colditz G A, Rosner B A, Speizer F E. J Natl Cancer Inst. 1996;88:365–371. doi: 10.1093/jnci/88.6.365. [DOI] [PubMed] [Google Scholar]