

Abstract
We have generated a mouse model for ataxia-telangiectasia by using gene targeting to generate mice that do not express the Atm protein. Atm-deficient mice are retarded in growth, do not produce mature sperm, and exhibit severe defects in T cell maturation while going on to develop thymomas. Atm-deficient fibroblasts grow poorly in culture and display a high level of double-stranded chromosome breaks. Atm-deficient thymocytes undergo spontaneous apoptosis in vitro significantly more than controls. Atm-deficient mice then exhibit many of the same symptoms found in ataxia-telangiectasia patients and in cells derived from them. Furthermore, we demonstrate that the Atm protein exists as two discrete molecular species, and that loss of one or of both of these can lead to the development of the disease.
Ataxia-telangiectasia (AT) is a human autosomal recessive disorder characterized by a wide variety of progressive clinical symptoms (for review, see ref. 1). The main symptoms of AT include cerebellar ataxia, oculocutaneous telangiectasias, acute sensitivity to ionizing radiation, thymic degeneration, immunodeficiencies and subsequent recurrent sinopulmonary infections, high incidences of lymphoma and leukemia, growth retardation, incomplete sexual maturation, and progeric changes to hair and skin. AT patients do not usually live past the second or third decade of life. Individuals heterozygous for AT mutations are spared most of the symptoms of the disease, but are at increased risk for developing malignancies (2, 3).
Cells derived from AT patients exhibit abnormal growth properties in culture (4–6). AT cells are chromosomally unstable, possess shortened telomeres, require higher serum concentrations, and exhibit reduced lifespan in culture. AT cells also exhibit abnormally high levels of spontaneous apoptosis and are remarkably sensitive to the effects of ionizing radiation and radiomimetic drugs. Induction of DNA damage by these agents fails to elicit a normal G1/S or G2/M arrest, resulting in radio-resistant DNA synthesis (for review, see refs. 6 and 7). Taken together, these findings suggest that an inability to respond normally to specific forms of radiation-induced DNA damage is a major cause of AT.
The gene mutated in AT was recently identified by positional cloning (8, 9). This gene, designated ATM, codes for a protein whose estimated molecular weight is 350 kDa. The ATM protein includes a phosphoinositol-3-kinase (PI-3-kinase)-like domain and a leucine zipper motif, indicating that its function may require interaction with DNA. This indication is further supported by the similarity between ATM and a number of molecules from other organisms, all of which participate in various aspects of cell-cycle control and maintenance of DNA integrity (for review, see ref. 10). A significant number of mutations, generally truncations, have been described in the ATM gene of AT patients (8, 11, 12).
Further research aimed at understanding the role of ATM and its involvement in cancer would benefit from the existence of a suitable animal model for AT. The nature of the mutations identified in the ATM gene and the high degree of similarity between the human and mouse gene products suggest that AT could be modeled by eliminating Atm function. We have therefore used gene targeting techniques to create a mouse strain that does not express the Atm protein. Mice lacking Atm are viable and display a series of pleiotropic defects that closely resemble those observed in AT patients. In this regard, the phenotype is similar to that very recently described in another atm-mutated strain of mice (13). In addition, we demonstrate that the ATM protein exists as two distinct molecular species, and that the clinical symptoms of AT can be elicited by the loss of one or both of these species.
MATERIALS AND METHODS
Construction of the ATM Targeting Vector.
A 226-bp fragment of the mouse atm cDNA was amplified by polymerase chain reaction from mouse spleen cDNA, using published sequence and expression data (8). The oligonucleotides used were derived from positions 5185–5207 (sense) and 5411–5391 (antisense) from the human ATM cDNA (ref. 9; GenBank accession no. U33841U33841). The amplified fragment was used to screen a mouse SV129 genomic library (λfixII, Stratagene), and a single clone from the mouse atm gene was isolated.
The targeting construct (Fig. 1B) was made by inserting a PGKneo cassette into a BamHI site contained within an exon present in the genomic clone, at a position corresponding to 5460 of the human ATM cDNA. In the final construct, prepared in the pPNT vector (15), this site was flanked by genomic fragments of 4.8 and 2.3 kb. The construct was linearized and electroporated into TC-1 embryonic stem cells (16). Following G418 and FIAU selection, 145 embryonic stem clones were isolated, and three of these were found to be correctly targeted.
Figure 1.
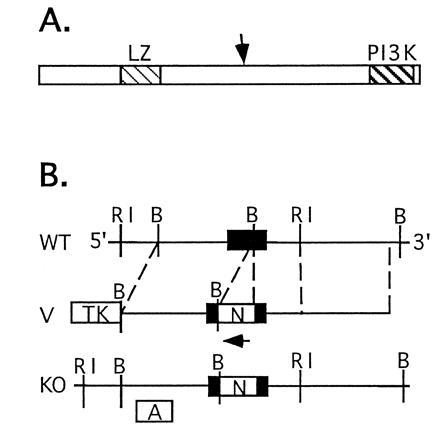
(A) Schematic drawing of the Atm protein based on ref. 14. Shown are the leucine zipper (LZ) and PI-3 kinase (PI3K) domains. An arrow marks the approximate location where the mutation described in the work was introduced. (B) Schematic drawing of the wild-type atm gene (WT), the targeting construct (V), and the mutated atm allele (KO). RI, EcoRI; B, BamHI. Note that the Neo and atm genes are transcribed in opposite orientations. Use of the internal probe A in conjunction with EcoRI digestion in DNA blot analysis results in bands of 6.5 kb and 8.3 kb for the wild-type and mutated alleles, respectively.
Cells from targeted clones were injected into C57BL/6J blastocysts, giving rise to a high-grade chimeric male mouse that passed the mutated atm allele to his offspring. The allele was carried on a mixed genetic background (129SvEv × Black Swiss). Genotyping was performed by DNA blot analysis (Fig. 2A), using a mouse atm cDNA probe corresponding to positions 4875–5263 of the human ATM cDNA in conjunction with EcoRI digestion.
Figure 2.
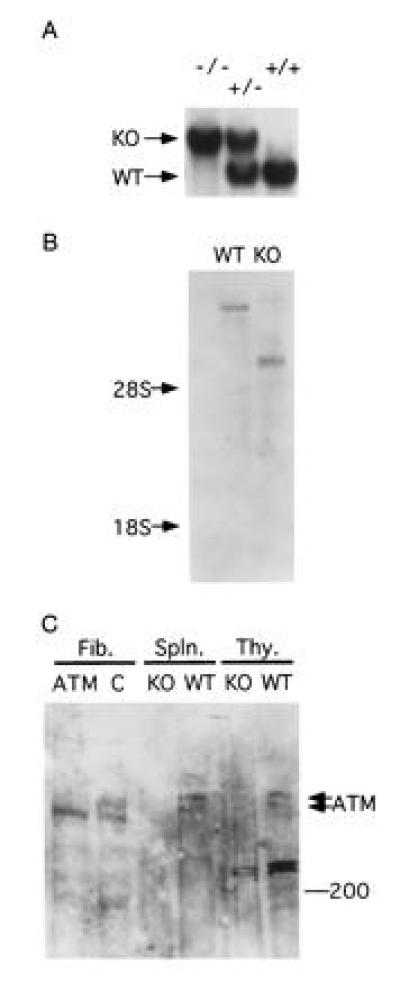
(A) Blot analysis of tail DNA prepared from wild type (+/+), Atm heterozygous (+/−), and homozygous (−/−) mutant mice. DNA was cut with EcoRI and probed with probe A (Fig. 1B). (B) Blot analysis of poly(A)+ selected RNA from thymi of wild-type (WT) and Atm-deficient (KO) mice. The blot was probed with a mouse Atm cDNA fragment as described. Each lane contained 2 μg of RNA. Equal loading was verified by ethidium bromide staining (not shown). (C) Blot analysis of proteins extracted from fibroblasts derived from healthy (C) or AT (ATM) humans and from thymi (Thy.) and spleens (Spln.) of wild-type and Atm-mutant mice.
RNA and Protein Analyses.
Blots were prepared from poly(A)+-selected RNA as described (17). The blots were probed with a fragment of the mouse atm cDNA corresponding to positions 104-5440 of the human ATM cDNA (A.E. and F.Z., unpublished results).
Protein extracts were prepared from thymi and spleens of adult male mice by crushing the organs in lysis buffer A (150 mM NaCl/50 mM Tris·HCl, pH 8.0/5 mM EDTA/1% Nonidet P-40/1 mM phenylmethylsulfonyl fluoride/0.1% SDS), supplemented with aprotonin (2 μg/ml), pepstatin (0.7 μg/ml), and leupeptin (0.5 μg/ml). Simian virus 40-transformed normal and AT human fibroblasts (repository nos. GM00637G and GM09607A, respectively) were obtained from the National Institute of General Medical Sciences Human Genetic Mutant Cell Repository (Camden, NJ). Cells were grown in Dulbecco’s modified Eagles’s medium (DMEM), supplemented with 10% fetal calf serum, 2 mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin, and were lysed in lysis buffer A. Protein (500 μg) was loaded from each sample onto a 1.5-mm thick SDS/5% polyacrylamide gel and electrophoresed at 4°C until the 200-kDa marker had traversed two thirds of the gel. Following semi-dry transfer onto an Immobilon P membrane (Millipore), the protein blot was processed, probed, and developed using Enhanced Chemiluminescence (Amersham) by standard techniques (18). The primary antibody used was polyclonal rabbit raised against a peptide corresponding to amino acids 13–24 of human ATM (9) (antibody PC85, Oncogene Research Products, Cambridge, MA).
Mouse Weight Measurements.
Mice were weighed at intervals of 3–4 days starting at weaning (19–21 days) and through adulthood (38–42 days). Same-sex littermates of all three genotypes were housed together in the same cages, with food and water provided ad libitum.
Histology.
Mouse tissue was removed and fixed in Optimal Fix (American Histology Reagent, Lodi, CA), blocked in paraffin, sectioned at 10 μm, and stained with hematoxylin and eosin.
Embryonic Fibroblast Culture and Karyotype Analysis.
Embryos (17.5 days old) were dissociated and plated in DMEM as described above and were subsequently genotyped by DNA blot analysis. For karyotype analysis, cells were grown on glass coverslips in RPMI 1640 medium supplemented with 10% fetal calf serum, 2 mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. When used, bleomycin (5 μg/ml, Calbiochem) was added 90 min before collection of cells. Colcemid (350 ng/ml, GIBCO/BRL) was then added to the growth medium and incubation was continued for 30 min. Medium was then replaced with 0.075 M KCl, followed by fixation in a 3:1 solution of methanol/acetic acid. For GTG banding, coverslips were dried overnight at 60°C, digested with 0.025% trypsin (GIBCO/BRL) in Gurr buffer, pH 7.0 (BDH) for 1 min, rinsed in the same buffer, and stained in Gurr Giemsa (BDH) for 5 min. Metaphase preparations were analyzed and photographed with a Zeiss Axiophot photomicroscope. Single chromatid breaks were scored as single events while double chromatid breaks were scored as two.
Flow Cytometry Analysis.
Samples were obtained from adult (>42 d) male mice. Cells prepared from spleen, thymus, or peripheral blood samples were stained with fluorescent antibodies and subjected to flow cytometry in a cytofluorograph II flow cytometer (Ortho Diagnostics). Data were analyzed with the cyclops software package (Cytomation, Ft. Collins, CO). The following monoclonal fluorescein isothiocyanate- or phycoerythrin-conjugated antibodies were used in this study: mouse anti-mouse thy1.1 (New England Nuclear), hamster anti-mouse CD3 and TCRαβ (clones 2c11 and H57, PharMingen) and rat anti-mouse CD4 and CD8 (clones L3T4 and 53–6.7, Becton Dickinson).
RESULTS
Generation of Atm-Deficient Mice.
The atm gene was disrupted by inserting a neo cassette within an exon at a site corresponding to nucleotide number 5460 of the human ATM (9) (Fig. 1). Following transfection, selection of targeted clones, and blastocyst microinjection, a high-grade chimeric male was produced and used to pass the Atm mutation through the germ line. F1 heterozygote mice were intercrossed, generating mice carrying no, one, or two copies of the mutant atm allele (Fig. 2A). Transmission of the mutant allele roughly followed a Mendelian inheritance pattern, and mice homozygous for the atm mutant allele were viable.
The ATM Protein Exists as Two Molecular Species That Are Absent from atm-Mutant Mice.
Examination of RNA purified from thymi of mutant atm homozygous mice indicated that the normal atm mRNA was replaced with two polyadenylylated mRNA species of abnormal size (Fig. 2B). Polyclonal antibodies raised against a peptide derived from the carboxy terminal region of human ATM protein were used to examine the status of the Atm protein in the mutant mice. Protein blots prepared from wild-type spleen and thymus revealed the presence of two closely spaced bands whose migration agreed with the expected size of the ATM protein, 350 kDa (Fig. 2C). Both bands were absent from organs of mice homozygous for the mutant atm allele, indicating that both were indeed products of the atm gene. The molecular basis for the existence of two Atm protein species is unknown at present. Further analysis failed to reveal smaller protein bands that could have represented truncated protein products of the disrupted atm gene (A.E., unpublished results). We therefore conclude that mice carrying the mutant atm fail to express the Atm protein product.
A similar pattern of two closely spaced bands was observed when protein extracts of virally transformed human fibroblasts were examined (Fig. 2C). Surprisingly, only the slower-migrating of the two bands was absent from the human AT fibroblasts. The precise molecular event underlying the AT phenotype in these cells is unknown and probably differs from the mutation introduced into the mouse gene. The data then indicate that the AT phenotype can be associated with the loss of either both or of the larger of the two protein species of ATM.
Atm Deficiency Causes Growth Retardation.
Mice homozygous for the atm mutation were found to be smaller in size and to weigh less than their wild-type or heterozygous littermates (Table 1 and Fig. 3). The difference was most striking at birth and at weaning (19–21 days of age), but remained clearly noticeable as the mice entered adulthood (38–42 days old). Although the growth curve of Atm-deficient mice appeared qualitatively normal, mutant mice generally remained the smallest of their litters even as adults (Fig. 3). The difference in body size was evident during nursing, when siblings compete for mother’s milk, as well as after weaning when food was freely available. This suggests that the growth defect observed is an intrinsic quality of these mice. In an analogous manner, embryo fibroblasts lacking Atm were more difficult to establish in culture and grew significantly slower than heterozygous or wild-type cells (data not shown).
Table 1.
Comparison of weights of Atm homozygous and control (wild type and Atm heterozygous) mice
| Age | Sex | Genotype | Weight ± SE | n | Change |
|---|---|---|---|---|---|
| 19–21 days | M | Control | 11.66 ± 0.18 | 54 | |
| M | Atm−/− | 9.17 ± 0.42 | 12 | −21% | |
| F | Control | 11.84 ± 0.24 | 45 | ||
| F | Atm −/− | 9.68 ± 0.46 | 12 | −18% | |
| 38–42 days | M | Control | 25.03 ± 0.34 | 32 | |
| M | Atm −/− | 21.40 ± 0.88 | 6 | −15% | |
| F | Control | 19.83 ± 0.49 | 17 | ||
| F | Atm −/− | 17.44 ± 0.75 | 8 | −12% |
Data are presented at weaning (19–21 days of age) and as adults (38–42 days of age). All values are statistically significant (P < 0.012) by Student’s t test.
Figure 3.
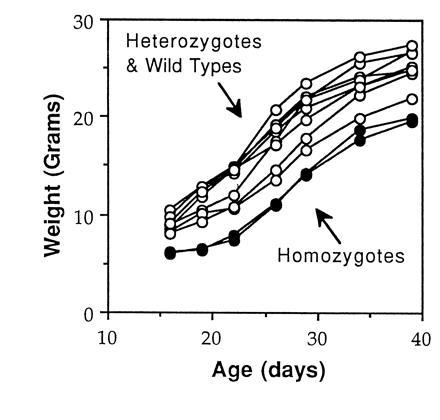
Atm-deficient mice are smaller than control littermates. The weight of male littermate mice of the indicated genotypes is shown as a function of time.
Atm-Deficient Fibroblasts Are Chromosomally Unstable.
To determine whether Atm deficiency was sufficient to cause chromosomal instability characteristic of AT, we examined metaphase chromosomes from normal and Atm-deficient embryo fibroblasts. Cells from Atm-deficient embryos exhibited nearly 6.5-fold more chromosomal breaks per metaphase than either of the other two genotypes (Table 2). No significant difference was noted between wild-type and Atm-heterozygous cells. Of note, this difference was detected in otherwise untreated cells, indicating that Atm deficiency leads to an inherent destabilization of chromosomal integrity.
Table 2.
Atm deficiency results in chromosomal instability
| Genotype | Breaks per metaphase (± SE) | n |
|---|---|---|
| +/+ | 0.71 ± 0.22 | 28 |
| +/− | 0.88 ± 0.28 | 17 |
| −/− | 4.56 ± 0.48 (× 6.4) | 52 |
| +/+, Bleomycin | 1.70 ± 0.39 | 20 |
| −/−, Bleomycin | 9.71 ± 0.99 (× 5.7) | 41 |
Metaphase chromosomes were prepared from embryonic fibroblasts of wild-type (+/+), heterozygous (+/−), or homozygous (−/−) Atm mutant mice, either with or without bleomycin treatment. Data are presented as the average of the number of double-stranded breaks observed per metaphase. The fold-increase in Atm homozygotes vs. wild-type controls is also noted.
Human AT fibroblasts and lymphocytes are known to be more susceptible than controls to the chromosome-damaging effects of the radiomimetic drug bleomycin (19–22). Treatment with bleomycin increased the number of chromosomal breaks observed in both wild-type and Atm-deficient cells (Table 2). However, the relative extent of this increase was similar in Atm-deficient and in wild-type cells. It appears that, under our experimental conditions, Atm-deficient cells are not more sensitive than controls to the DNA-damaging effects of bleomycin.
Aspermiogenesis in Atm-Deficient Mice.
To examine the effects of Atm deficiency on reproductive capability, we examined the reproductive system of several Atm-deficient mice. Testes of Atm-deficient mice were markedly smaller and structurally abnormal when compared with controls. Seminiferous tubules of Atm-deficient mice were composed of normal-looking basement membrane and Sertoli cells (Fig. 4B), which normally support the developing spermatozoa. Germ cells that had undergone mitosis were observed in some of the seminiferous tubules. However, no evidence of further development was observed. This resulted in a total lack of maturing sperm within the tubules and of mature sperm in the tubule lumen (Fig. 4B), the epididymis, and ductus deferens (not shown). We conclude that Atm function is required for spermatogonia to develop into mature sperm. In a manner consistent with this result, attempts to mate Atm-deficient male mice with wild-type females have been unsuccessful.
Figure 4.
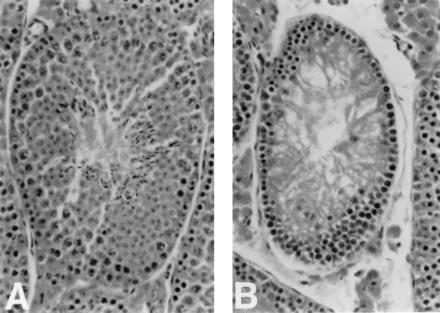
Cross section through a seminiferous tubule of a wild-type (A) and an Atm-deficient (B) adult mouse. Note that the developing sperm present in A is replaced by debris in B. (×200.)
T Cell Deficiencies and Thymomas in Mice Lacking Atm.
To determine whether Atm deficiency results in the severe immunological abnormalities associated with AT, the immune system of Atm-deficient mice was examined. Thymi from Atm-deficient mice were found to be significantly smaller and consistently yielded fewer cells upon disaggregation when compared with organs from wild-type mice. Upon histological examination, Atm-deficient thymi were found to have a reduced cortex and prominent medulla (data not shown).
To further study T-cell development in Atm-deficient mice, samples of mouse thymus, spleen, and peripheral blood were analyzed by flow cytometry (Table 3). Although their relative proportion of Thy-1-positive cells was unchanged, thymi from Atm-deficient mice exhibited a fourfold reduction in cells expressing either CD3 or αβTCR. Thymi from Atm-deficient mice exhibited an increase in CD4+/CD8+ double positive cells, concomitant with a fourfold decrease in CD4+/CD8− single positive cells. A slight decrease in CD4−/CD8+ single-positive cells was noted but was not statistically significant. Overall, it appears that Atm function is needed in the thymus to ensure correct transition of T cells from the CD4+/CD8+ double-positive stage to the mature single-positive stage.
Table 3.
Reduced T cell population in thymus, spleen, and peripheral blood from mutant Atm homozygous mice
| Tissue | Genotype | Thy1 | CD3 | αβTCR | CD4−CD8− | CD4+CD8− | CD4−CD8+ | CD4+CD8+ |
|---|---|---|---|---|---|---|---|---|
| Thymus | Control | 98.8 ± 0.6 | 20.3 ± 1.5 | 18.0 ± 1.7 | 2.8 ± 1.6 | 12.1 ± 0.5 | 8.9 ± 1.6 | 78.0 ± 2.3 |
| Atm | 98.5 ± 0.4 | 5.0 ± 1.3* | 5.4 ± 1.3* | 2.9 ± 0.9 | 3.6 ± 0.9* | 4.6 ± 1.4 | 88.9 ± 3.0** | |
| Spleen | Control | 33.7 ± 1.7 | 41.6 ± 1.4 | 44.7 ± 1.7 | 60.2 ± 1.9 | 25.0 ± 1.6 | 14.5 ± 0.7 | 0.2 ± 0.1 |
| Atm | 10.0 ± 1.5* | 15.6 ± 1.9* | 20.0 ± 1.8* | 85.5 ± 1.8* | 8.8 ± 1.2* | 5.4 ± 1.4* | 0.2 ± 0.1 | |
| Blood | Control | 39.7 ± 4.6 | 75.4 ± 3.1 | ND | 28.3 ± 2.1 | 54.9 ± 1.7 | 16.6 ± 0.8 | 0.2 ± 0.1 |
| Atm | 15.9* | 34.6* | ND | 45.1** | 37.3* | 17.6 | 0.1 |
Shown are the percentage of cells staining positive for each surface molecule by flow cytometry in Atm homozygotes and control (Atm heterozygote and wild-type) mice. n = 3 in all samples except in ATM blood, where n = 2. ND, not determined. ∗, P < 0.01; ∗∗ 0.01 < P < 0.05 by Student’s t test analysis.
Flow cytometry analysis of spleens from Atm-deficient mice revealed two- to threefold reductions in cells expressing the T-cell-specific markers Thy-1, CD3, αβTCR, CD4, or CD8 (Table 3). The relative proportion of cells expressing the B-cell-specific markers B220 or IgM was not altered significantly (data not shown). Similar results were obtained with peripheral blood (Table 3). Taken together, it appears that Atm deficiency has a profound and selective negative effect on T cell development. Nonetheless, four of the ATM-deficient mice have gone on to develop malignant thymomas between 3 and 4 months of age.
Spontaneous Apoptosis in Cultured Atm-Deficient Thymocytes.
The defects found in the number and characteristics of T cells in Atm-deficient mice prompted us to examine whether these cells were flawed also in their response to ionizing radiation. Freshly isolated thymocytes from Atm-deficient mice and from controls were therefore irradiated with doses of x-rays ranging from 0.1 to 20 Gy. Following an 8-hr in vitro recovery period, the extent of radiation-induced apoptosis was examined by DNA laddering and by flow cytometry analysis. Preliminary experiments revealed that the extent of apoptosis caused by a given dose of radiation was similar in ATM-deficient and in control thymocytes (not shown).
Interestingly, unirradiated thymocytes lacking Atm appeared to undergo significantly more apoptosis following in vitro incubation than did control cells (Fig. 5). This difference, which was reproducible, was a direct result of in vitro incubation, as the extent of apoptosis in freshly isolated thymocytes from Atm-deficient and from control mice was similar (Fig. 5). Further work is needed to determine whether this phenomenon is caused by inflexible requirements for a particular growth factor or by another mechanism. In any case, a similar situation in vivo would act to reduce thymocyte populations and could account for thymic atrophy of AT patients.
Figure 5.
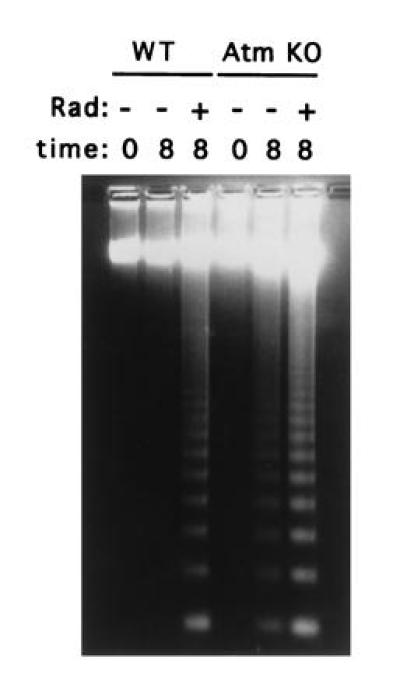
DNA laddering in thymocytes of wild-type and Atm-deficient mice. Freshly isolated thymocytes were plated after either no irradiation (−) or irradiation at 3.5 Gy (+). DNA was prepared either immediately afterward (0) or after 8 hr incubation (8), and 10 μg was electrophoresed through a 1% agarose gel.
DISCUSSION
We have created a strain of mice in which the atm gene has been inactivated and gives rise to no protein product. Atm-deficient mice exhibit many of the pleiotropic symptoms that are found in AT patients. Mice lacking Atm are smaller than their normal littermates, suffer from abnormalities in their T cell compartment, and are infertile. Cells derived from Atm-deficient mice grow poorly in culture, are genetically unstable, and appear to undergo spontaneous apoptosis more readily than control cells. In a manner analogous to the human disease, mice heterozygous for the mutant atm allele are generally free of these symptoms.
Several of our results indicate that Atm plays a major role in promoting growth. Mice deficient for Atm are born small and remain smaller than their normal or heterozygous littermates. The intrinsic nature of this defect is supported also by the findings that Atm-deficient fibroblasts grow poorly in culture and by the atrophy of the thymus and testes in these animals. Growth retardation and atrophy of gonads, thymus, thyroid, and adrenal gland have been described in AT (1), indicating that a similar intrinsic mechanism may be acting in human patients.
Genetic instability, which is commonly observed in AT (1, 6), can account for the observed growth retardation. ATM is believed to activate a number of cellular functions aimed at promoting cell survival in the face of DNA damage (6). Acting in a concerted manner, these functions halt p53-mediated programmed cell death and up-regulate the various cell-cycle checkpoints and DNA repair mechanisms. Lack of ATM function would then be expected to favor apoptosis under circumstances where DNA repair would normally occur. Indeed, cells from AT patients exhibit high basal levels of DNA damage and reduced thresholds for apoptosis (ref. 23; reviewed in ref. 6). We have demonstrated that fibroblasts from Atm-deficient mice exhibit a sixfold increase in double-stranded chromosome breaks, indicating high basal levels of DNA damage. Furthermore, increased levels of spontaneous apoptosis observed in cultured Atm-deficient thymocytes argue that these cells have a lower threshold for survival than control cells. Widespread, ongoing apoptosis could be expected to result in loss of body mass and retarded growth of AT patients and Atm-deficient mice.
Of note, cells that have both increased levels of DNA damage and a lower threshold for apoptosis in response to it are expected to be effectively eliminated. However, cells with high basal levels of DNA damage are a consistent finding in AT and in Atm-deficient mice. It is then possible that cell types severely depleted from Atm-deficient mice, such as maturing sperm, are those that combine increased levels of DNA damage with lowered apoptotic thresholds. On the other hand, Atm deficiency in other cell types, such as cultured fibroblasts, may lower their capability to repair DNA without a concomitant increase in sensitivity to apoptosis. The functions of Atm with respect to apoptotic thresholds and DNA damage repair then appear to be regulated independently of one another and in a cell-specific manner.
T lymphocyte development and function are known to be abnormal in AT. Decreases in mature T-cells expressing the αβ T cell receptor or CD4 have been reported in AT patients (24, 25), as have defects in T cell signaling (26). Abnormalities in T cell receptor gene recombination, which can lead to the development of T-cell tumors, have also been described in AT (for reviews, see refs. 27 and 28). Atm-deficient mice have significantly reduced amounts of mature circulating T cells. Analysis of maturing T cells in the thymus indicates a decrease in mature CD4 or CD8 single positive cells concomitant with an increase in CD4+/CD8+ double-positive cells. Atm-deficient thymocytes are also less capable of withstanding transfer to culture conditions, indicating a possible fragility in vivo as well. Interestingly, B cells are known to be less affected than T cells in AT (28), and we have found this to be the case in Atm-deficient mice as well. Taken together, these results indicate that Atm deficiency is sufficient to cause the major T cell defects observed in AT.
Recently, an account of another mouse model for AT has been published (13). Most of the aspects of the AT phenotype that were examined in both lines of mice yielded identical results. In particular, mice from both lines exhibit growth retardation, thymic atrophy, male sterility, and decreased T-cell and unaffected B-cell populations, and yield fibroblasts that grow poorly in culture. This similarity indicates that, as in the case of the human disease, different mutations in the atm gene can yield grossly similar phenotypes. We have further demonstrated that Atm-deficient fibroblasts are genetically unstable and that Atm-deficient thymocytes undergo spontaneous apoptosis when cultured. We note that Barlow et al. (13) have observed thymic lymphoblastic lymphomas in their atm-mutant mice that arise between 2–4 months of age. Four of twelve of our Atm-deficient mice have also developed thymomas, the earliest tumor occurring at 3 months. Not all AT patients develop lymphoma and there is a degree of variation in the nature of the disease in those that do (for review, see ref. 28).
Finally, the Atm and ATM proteins are detected as two protein species on protein blots. Both molecular species are absent from Atm-deficient mice, indicating that both are indeed derived from the atm gene. A human AT cell line was found to have lost only the slower-migrating of the two ATM bands. This may indicate that AT can result from elimination of only one of the two molecular species of ATM. It remains to be seen whether loss of the faster-migrating molecular species also leads to AT. The molecular basis for the existence of the two molecular species of ATM is unknown at present. It is possible that the two bands represent polypeptides that have undergone different posttranslational modifications. Alternatively, the atm and ATM genes may have more than one promoter and may give rise to alternate isoforms. Further characterization of the atm gene and of the pattern of Atm protein expression are required to clarify this issue.
Acknowledgments
We thank Dr. Robert Cardiff and his staff of the Transgenic Mouse Pathology Laboratory (University of California School of Medicine, Davis) for preparation and interpretation of the histological sections. We also thank Anne Piecewicz and W. Lee Powell (Department of Pathology, Brigham and Women’s Hospital) for help with banding and photomicroscopy of metaphase chromosomes.
Footnotes
Abbreviations: AT, ataxia-telangiectasia; ATM and atm, mutated AT gene from human and mouse, respectively; ATM and Atm, AT protein from human and mouse, respectively.
References
- 1.Sedgewick R, Boder E. In: Handbook of Clinical Neurology. Vinken P, Bruyn G, Klawans H, editors. New York: Elsevier; 1991. pp. 347–423. [Google Scholar]
- 2.Swift M, Reitnauer P J, Morrell D, Chase C L. N Engl J Med. 1987;316:1289–1294. doi: 10.1056/NEJM198705213162101. [DOI] [PubMed] [Google Scholar]
- 3.Easton, D. F. (1994) Int. J. Radiat. Biol. 66, Suppl., S177–S182. [DOI] [PubMed]
- 4.Metcalf J A, Parkhill J, Campbell L, Stacey M, Biggs P, Byrd P J, Taylor A M R. Nat Genet. 1996;13:350–353. doi: 10.1038/ng0796-350. [DOI] [PubMed] [Google Scholar]
- 5.Pandita T K, Pathak S, Geard C R. Cytogenet Cell Genet. 1995;71:86–93. doi: 10.1159/000134069. [DOI] [PubMed] [Google Scholar]
- 6.Meyn M S. Cancer Res. 1995;55:5991–6001. [PubMed] [Google Scholar]
- 7.Shiloh Y. Eur J Hum Genet. 1995;3:116–138. doi: 10.1159/000472285. [DOI] [PubMed] [Google Scholar]
- 8.Savitsky K, Bar-Shira A, Gilad S, Rotma G, Ziv Y, et al. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 9.Savitsky K, Sfez S, Tagle D A, Ziv Y, Sartiel A, Collins F S, Shiloh Y, Rotman G. Hum Mol Genet. 1995;4:2025–2032. doi: 10.1093/hmg/4.11.2025. [DOI] [PubMed] [Google Scholar]
- 10.Zakian V A. Cell. 1995;82:685–687. doi: 10.1016/0092-8674(95)90463-8. [DOI] [PubMed] [Google Scholar]
- 11.Byrd P J, McConville C M, Cooper P, Parkhill J, Stankovic T, McGuire G M, Thick J A, Taylor A M R. Hum Mol Genet. 1996;5:145–149. doi: 10.1093/hmg/5.1.145. [DOI] [PubMed] [Google Scholar]
- 12.Gilad S, Khosravi R, Uziel T, Ziv Y, Rotman G, et al. Hum Mol Genet. 1996;5:433–439. doi: 10.1093/hmg/5.4.433. [DOI] [PubMed] [Google Scholar]
- 13.Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley J N, Ried T, Tagle D, Wynshaw-Boris A. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 14.Pecker I, Avraham K B, Gilbert D J, Savitsky K, Rotman G, Harnik R, Fukao T, Schrock E, Hirotsune S, Tagle D, Collins F S, Wynshaw-Boris A, Reid T, Copeland N G, Jenkins N A, Shiloh Y, Ziv Y. Genomics. 1996;35:39–45. doi: 10.1006/geno.1996.0320. [DOI] [PubMed] [Google Scholar]
- 15.Tybulewicz V L, Crawford C E, Jackson P K, Bronson R T, Mulligan R C. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 16.Deng C, Wynshaw-Boris A, Kuo A, Zhou F, Leder P. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- 17.Elson A, Deng C, Campos-Torres J, Donehower L A, Leder P. Oncogene. 1995;11:181–190. [PubMed] [Google Scholar]
- 18.Harlow E, Lane D. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. [Google Scholar]
- 19.Cohen M M, Simpson S J, Pazos L. Cancer Res. 1981;41:1817–1823. [PubMed] [Google Scholar]
- 20.Kohn P H, Kraemer K H, Buchanan J K. Exp Cell Res. 1982;137:387–395. doi: 10.1016/0014-4827(82)90040-4. [DOI] [PubMed] [Google Scholar]
- 21.Shaham M, Becker Y, Lerer I, Voss R. Cancer Res. 1983;43:4244–4247. [PubMed] [Google Scholar]
- 22.Taylor A M R, Rosney C M, Campbell J B. Cancer Res. 1979;39:1046–1050. [PubMed] [Google Scholar]
- 23.Duchaud E, Ridet A, Stoppalyonnet D, Janin N, Moustacchi E, Rosselli F. Cancer Res. 1996;56:1400–1404. [PubMed] [Google Scholar]
- 24.Carbonari M, Cherchi M, Paganelli R, Giannini G, Galli E, Gaetano C, Papetti C, Fiorilli M. N Engl J Med. 1990;322:73–76. doi: 10.1056/NEJM199001113220201. [DOI] [PubMed] [Google Scholar]
- 25.Paganelli R, Scala E, Scarselli E, Ortolani C, Cossarizza A, Carmini D, Aiuti F, Fiorilli M. J Clin Immunol. 1992;12:84–91. doi: 10.1007/BF00918137. [DOI] [PubMed] [Google Scholar]
- 26.Kondo N, Inoue R, Nishimura S, Kasahara K, Kameyama T, Miwa Y, Lorenzo P R, Orii T. Scand J Immunol. 1993;38:45–48. doi: 10.1111/j.1365-3083.1993.tb01692.x. [DOI] [PubMed] [Google Scholar]
- 27.Kirsch, I. R. (1994) Int. J. Radiat. Biol. 66, Suppl., S97–S108. [PubMed]
- 28.Taylor A M, Metcalfe J A, Thick J, Mak Y F. Blood. 1996;87:423–438. [PubMed] [Google Scholar]