

Abstract
Early in ontogeny, the secondary lymphoid organs become populated with numerous cells of mesodermal origin which forms both the lymphoid and stromal elements. The critical receptor/ligand interactions necessary for lymphoid organogenesis to occur are for the most part unknown. Although lymphotoxin-α (LTα) has been shown to be required for normal lymph node, Peyer’s patch, and splenic development, it is unclear if soluble LTα3, and/or cell-bound lymphotoxin-αβ (LTαβ) mediate these developmental events. Here we report that blocking LTαβ/lymphotoxin-β receptor (LTβR) interaction in vivo by generating mice which express a soluble LTβR–Fc fusion protein driven by the human cytomegalovirus promoter results in an array of anatomic abnormalities affecting both the spleen and Peyer’s patches, but not the lymph nodes. These results demonstrate that surface LTαβ ligand plays a critical role in normal lymphoid organ development.
Tumor necrosis factor (TNF), lymphtoxin α (LTα), and lymphotoxin-β (LTβ) are related cytokines which belong to the TNF ligand family and are encoded by genes clustered within the major histocompatibility complex gene complex (1). Both TNF and LTα self-associate into structurally related homotrimers that bind the same receptors, the p55–60-kDa receptor (type 1 or CD120a; TNFRp60) and the p75–80-kDa receptor (type 2 or CD120b; TNFRp80) (2). TNF is expressed in both cell-bound and soluble forms, while LTα3 is only produced as a secreted molecule. LTβ, on the other hand, exists as a membrane-bound heterotrimeric complex in association with LTα forming two complexes, LTα1β2, and LTα2β1 (1). LTα1β2 is the major cell surface complex expressed only on lymphoid cells, and binds the lymphotoxin-β receptor (LTβR) expressed on nonlymphoid cells (1, 3, 4).
Human and murine TNF and human LTα3 have been extensively characterized and have been shown to induce many of the same functions. Thus, it has been suggested that these cytokines are redundant. Little is known about the murine LTα3 and LTαβ ligands, largely due to the lack of specific reagents. However, recent reports have demonstrated that mice lacking LTα expression by selective gene targeting have disorganized splenic architecture and lack lymph nodes and Peyer’s patches, while the thymus is unaffected (5, 6). Since normal lymph node development occurs in TNFRp60- and TNFRp80-deficient mice, or in mice expressing a soluble TNFRp60–Fc transgene, a crucial role for LTαβ ligand in these processes is likely (7–10). To directly test if surface LTαβ is required for normal lymphoid organogenesis to occur, we neutralized LTαβ/LTβR interactions in vivo by generating mice which constitutively express a soluble murine LTβR-human IgG1 (LTβR–Fc) transgene, whose expression is driven by the human cytomegalovirus (CMV) promoter. The human IgG1 Fc component was specifically mutated to inhibit FcR binding and complement fixation. Our results demonstrate that surface LTαβ is a crucial ligand for normal splenic and Peyer’s patch development.
MATERIALS AND METHODS
Plasmid Construction.
To generate soluble LTβR–Fc chimeric protein, the extracellular domain of murine LTβR and a mutated human IgG1 Fc (kindly provided by C. Ambrose, Biogen) were used (11). To inhibit FcR binding and complement fixation, the human IgG1 Fc was mutated at the CH2 domain (L234A, L235E, G237A, and P331S) (12–14). The extracellular domain of the LTβR and the mutated hIgG1 were isolated as a 0.69-kb NotI/SalI fragment, and a 0.67-kb SalI/NotI fragment, respectively. The two DNA fragments were then ligated into the NotI site of the CA116 vector containing the human CMV promoter (−553 to +71) (CA116, a derivative of SAB132 was provided by C. Ambrose) (15). The CMV–LTβR–Fc transgene was then isolated as an EcoRI/HindIII fragment, and purified with a QIAquick gel extraction kit (Qiagen, Chatsworth, CA) following manufacturer’s instructions.
Transgenic Mouse Production and Detection.
The purified CMV–LTβR–Fc transgene (2–10 ng/μl) was microinjected into BALB/c-fertilized oocytes from FVB mice, and implanted into pseudopregnant (BALB/c × 129)F1 mice in a Stanford transgenic mouse facility. All mice were bred and maintained at the Stanford University animal facility under barrier isolation conditions. Genomic DNA was isolated from mice tails biopsied at 3 weeks of age, and offspring containing the integrated transgene were identified by Southern blot analysis using PstI digested genomic DNA. Soluble chimeric protein in the sera was quantitated by ELISA: 96-well polyvinyl microtiter dishes were coated with 10 μg/ml of purified goat anti-human antibody (Sigma), the sera were then titered onto the treated plates and detected with alkaline phosphate-conjugated goat anti-human Fc polyclonal antibody (Caltag, South San Francisco, CA) and substrate [p-nitrophenyl phosphate (PNPP) in DEA buffer]. Optical density was quantified at 420 nM. Transgene positive mice were then crossed to BALB/c mice to establish founder lines, from which heterozygous mice were intercrossed.
Immunohistology.
All mice, from two separate founder lines (1610 and 201), were weighed and spleens, lymph nodes, Peyer’s patches, and thymi were removed from age and sex matched transgenic-positive and -negative mice. When transgenic-negative littermates were not available, age and sex matched BALB/c mice were used as control mice. Lymphoid organs were excised and either: fixed in formalin, embedded in paraffin, and stained with hematoxylin/eosin or embedded in O.C.T. (Miles) compound and frozen in liquid nitrogen. Frozen tissue sections were fixed in ice-cold acetone for 10 min and double stained for T- and B-cell surface antigens with phycoerythrin-conjugated anti-CD4 (PharMingen) and biotinylated anti-B220 (PharMingen), followed by fluorescein-conjugated avidin (Neutralite, Southern Biotechnology Associates). The combined T- and B-cell images were digitized and processed as described (16). Frozen tissue sections were also stained with antibodies specific for reticular fibroblasts (ER-TR7), marginal zone macrophages (ER-TR9), marginal zone metallophilic macrophages (MOMA-1), or MAdCAM-1 vascular addressin (MECA367) as described (17). The antibodies were detected with anti-rat immunoglobulin-conjugated with horseradish peroxidase (Dako), followed by incubation with diaminobenzidene (Sigma). In data not shown, mesenteric lymph nodes were stained with antibodies to MAdCAM-1 vascular addressin on high endothelial venules (MECA367), and macrophages (ER-TR9, and MOMA-1), and thymi were stained with antibodies specific for cortical epithelium (ER-TR4), medullary epithelium (ER-TR5), fibroblasts (ER-TR7), and dendritic cells (N418) (18–20).
Specificity and Bioactivity of the LTβR–Fc Fusion Protein.
The capacity of the LTβR–Fc fusion protein present in the sera to neutralize LTαβ-mediated cytotoxicity was determined with the murine WEHI-164 fibrosarcoma cycloheximide (CHX) methyl tetrazolium salt (MTT) cytotoxicity assay as described (21). Briefly, 1 × 104 cells were incubated overnight in 96-well plates. Titrations of purified LTαβ were added to the cells in the presence of 10 μg/ml CHX and either 1% normal mouse sera (NMS), 1% sera isolated from the transgenic-positive mice, or 10 ng/ml of purified control LTβR–Fc fusion protein in the presence of 1% NMS. Cell viability was determined 16 hr later by MTT (Sigma). Relative inhibition was determined by the concentration of LTαβ ligand which was required for 50% cytolysis of the target population in the presence of 1% serum from transgenic-positive mice relative to the amount of ligand which gave 50% cytolysis in the presence of 1% NMS. Specificity of the LTβR–Fc transgene was determined by the inability of 1% sera from transgenic-positive mice to inhibit either TNF or LTα- mediated killing of the WEHI-164 cells. The recombinant forms of murine LTα and murine LTαβ were prepared as described for the human ligands and shown to signal through the TNFRp60 and LTβR, respectively, in this assay system (unpublished data).
RESULTS
To test the role of surface LTαβ in normal lymphoid development and function, we blocked LTαβ/LTβR interactions in vivo by generating mice which constitutively express a soluble murine LTβR–human IgG1 (Fc) transgene driven by the CMV promoter. The Fc portion of the transgene was specifically mutated to inhibit FcR binding and complement fixation to avoid depletion of LTαβ-expressing cells in vivo. The lack of FcR interactions by the mutated IgG1 chimeric protein were previously verified in FcR binding assays (22). The LTβR–Fc chimeric construct was put under the control of the CMV promoter to achieve systemic expression of the soluble fusion protein in vivo. The CMV promoter has been reported to be transcriptionally active early in ontogeny and expressed in most organs including thymus, spleen, bone marrow, and stomach (23, 24).
The purified LTβR–Fc fusion construct was microinjected into BALB/c-fertilized FVB oocytes, and mice with an integrated transgene were identified by Southern blot analysis. Soluble chimeric protein activity in the sera was quantitated by ELISA as described in Materials and Methods. From 11 mice which integrated the LTβR–Fc transgene, 4 separate founder lines were established, 2 of which expressed high circulating levels of the fusion protein in the sera (≈1.5 μg/ml), and 2 of which expressed low serum levels (≈50–100 ng/ml). The level of soluble fusion protein in the sera detected by ELISA correlated directly with the ability of the circulating chimeric protein to specifically inhibit LTαβ- but not LTα3- or TNF-α-mediated killing of WEHI-164 cells (Table 1). Offspring derived from the founders which expressed low serum levels of the fusion protein, consistently also expressed low circulating levels. Conversely, mice derived from founders which expressed high transgene levels were found to have a large variation in circulating LTβR–Fc fusion protein (0–1.5 μg/ml). Heterozygous mice expressing the highest levels of the LTβR–Fc fusion protein were intercrossed to obtain mice which expressed up to 6.5 μg/ml of the chimeric protein in the sera. However, circulating LTβR–Fc fusion protein did not reach neutralizing levels (based on in vitro assays) until 3 days after birth; newborn pups had undetectable fusion protein in their sera. Circulating LTβR–Fc fusion protein dropped from high to almost undetectable levels during pregnancy. Thus, placental transfer of the LTβR–Fc protein was presumably not sufficient to neutralize surface LTαβ ligand in the developing fetus.
Table 1.
Specificity and dose-dependency of the LTβR–Fc chimeric fusion protein
| Mouse* | Tg† | LTβR–Fc‡, μg/ml | Relative inhibition of cytokine
cytotoxicity§
|
Body, g | Spleen, g | Thymus, g | Peyer’s patches
|
|||
|---|---|---|---|---|---|---|---|---|---|---|
| LTαβ | LTα | TNF | n | cm2 | ||||||
| 1610.20¶ | + | 0.05 | 1.1 | <1 | <1 | 19.5 | 0.093 | 0.128 | 11 | 0.35 |
| 1610.30 | + | 0.10 | 1.6 | <1 | <1 | 20.3 | 0.084 | 0.097 | 8 | 0.31 |
| 1610.1 | + | 0.50 | 6.7 | <1 | <1 | 19.8 | 0.078 | 0.121 | 10 | 0.15 |
| 1610.3 | + | 0.50 | 5.8 | <1 | <1 | 17.1 | 0.055 | 0.089 | 7 | 0.16 |
| 1610.10 | + | 1.25 | 18.3 | <1 | <1 | 17.8 | 0.055 | 0.084 | 5 | 0.09 |
| 1610.2 | + | 1.30 | 19.2 | <1 | <1 | 14.5 | 0.049 | 0.113 | 5 | 0.07 |
Mice were 6-week-old female littermates born from heterozygous parents.
Expression of the LTβR–Fc transgene was determined by Southern blot analysis.
Concentration of the soluble LTβR–Fc fusion protein in the sera was determined by ELISA.
Determined by the ability of 1% sera to inhibit either LTαβ-, LTα-, or TNF-mediated killing of WEHI-164 cells; 10 ng/ml of purified murine LTβR–Fc fusion protein in 1% NMS was able to inhibit LTαβ-killing by 19.2-fold.
Similar values were observed with age- and sex-matched nontransgenic mice.
Although mice which expressed low levels of the soluble LTβR–Fc fusion protein appeared phenotypically and histologically normal, correlating with the low ability of the sera to neutralize LTαβ activity in vitro (Table 1), mice which expressed high levels of the chimeric protein had many immunologic abnormalities. Offspring derived from the same high-expressor founder line had a large variation in transgene product expression, resulting in wide phenotypic variation. Many of the mice expressing high circulating LTβR–Fc protein levels had reduced body and spleen weight compared with their nontransgenic littermates. Peyer’s patches were reduced in size or totally absent, and large variations in thymus size were observed (Fig. 1). The reduction of spleen weight and Peyer’s patch size correlated directly with circulating LTβR–Fc protein levels in most mice examined. Conversely, thymus size and transgene expression appeared not to be correlated (Table 1). All transgenic mice with low body weight had high serum levels of the LTβR–Fc chimeric protein, however, not all mice with high circulating LTβR–Fc levels had low body weight.
Figure 1.
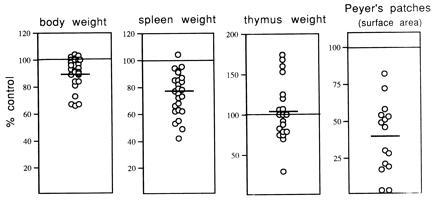
Reduced size of spleen and Peyer’s patches in mice expressing a soluble LTβR–Fc transgene. Three- to 15-week-old mice expressing high levels of LTβR–Fc transgene product in the sera (0.5–6.5 μg/ml) from two separate founder lines were weighed and sacrificed, and the weight of the thymi and spleens was determined. Small intestine Peyer’s patches were counted, and the serosal surface area was determined. The percent control was determined by directly comparing sex-matched nontransgenic littermates to transgene-positive mice. Horizontal lines represent the group averages, and the mean ± the SD are as follows: body weight, 91% ± 11.0 (40 mice from 10 separate litters); spleen weight, 75% ± 15.7 (35 mice from 9 separate litters); thymus weight, 103% ± 35.2 (34 mice from 8 separate litters); and Peyer’s patch area, 39% ± 23.9 (23 mice from 6 separate litters).
Unexpectedly, lymph nodes were present in all mice examined, and appeared phenotypically normal with respect to T cell and B cell surface antigens (data not shown). Circulating LTβR–Fc transgene levels were undetectable at birth, and did not reach neutralizing levels until after day 3 of life (data not shown). Rennert et al. (16) have recently demonstrated that soluble LTβR–Fc was able to inhibit lymph node development in newborn mice if administered to the pregnant mothers during days 11–16 of gestation; lymph node development occurred normally if the fusion protein was administered after day 16 of gestation. Thus, it appears that lymph node development is occurring in our mice due to late onset expression of the LTβR–Fc transgene.
Expression of the transgene did not appear to affect lymph node or thymic structure. Brachial, axillary and popliteal lymph nodes, stained with hematoxylin/eosin, showed normal architecture in mice expressing both low and high levels of the LTβR–Fc fusion protein (Fig. 2 A and B). Segregation between the T-cell-rich paracortex, and the B-cell-rich primary follicles was also observed in these mice (Fig. 2 C and D). High endothelial venules and macrophages of mesenteric lymph nodes appeared normal, in both their presence and distribution, as determined by immunohistology (data not shown). Even though the Peyer’s patches were reduced (or absent) both in size and number in mice expressing high levels of the LTβR–Fc fusion protein, the Peyer’s patches which did develop appeared normal, with visible germinal centers (Fig. 2 E and F). Both the medullary and cortical zones of the thymus were unaffected by transgene expression, as demonstrated by hematoxylin/eosin staining and immunohistology using antibodies specific for cortical epithelium, medullary epithelium, dendritic cells, and fibroblasts (data not shown). The ratio of single-positive and double-positive thymocytes isolated from “high-expressing” mice was indistinguishable from that of the transgenic-negative littermates. Blood isolated from these mice was normal with respect to hemoglobin, hematocrit, as well as leukocyte and platelet counts, compared with nontransgenic littermates. Bone marrow also appeared phenotypically normal.
Figure 2.
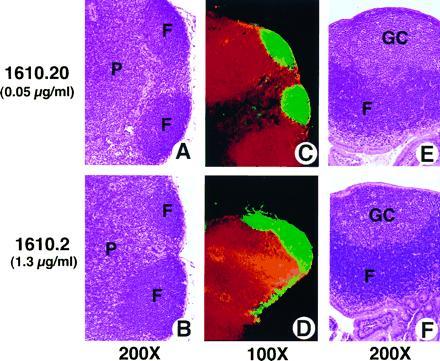
Normal architecture of lymph nodes and Peyer’s patches from LTβR–Fc-expressing mice. Tissue sections of lymph node (A–D) and Peyer’s patch (E and F) from either low-expressing (A, C, and E), or high-expressing (B, D, and F) 6-week-old female LTβR–Fc transgenic mice (1610.2 and 1610.20 from Table 1). Tissue sections were stained with either hematoxylin/eosin (A and B, E and F) (×200), or with antibodies specific for T-cell (anti-CD4, red) and B-cell (anti-B220, green) antigens (C and D) (×100). Yellow staining indicates colocalization of both T and B cells. F, P, and GC indicate the location of the B-cell-rich follicles, the T-cell-rich paracortex, and the germinal centers, respectively.
However, histologic and immunohistologic analysis of mice from 13 separate litters, derived from two separate founder lines (1610 and 201), showed a specific and dose-dependent effect of LTβR–Fc transgene expression on splenic development. In all mice expressing high serum levels of the LTβR–Fc fusion protein (0.5–6.5 μg/ml), a dramatic disruption of splenic architecture was observed. As circulating LTβR–Fc expression increased, a progressive loss of splenic marginal zones, as well as T-cell/B-cell organization were found. Specifically, hematoxylin/eosin stained tissue sections showed that in mice expressing high, but not low, levels of the LTβR–Fc transgene, the total volume of the white pulp was decreased, with condensation of the peri-arteriolar-lymphoid sheath (PALS) (Fig. 3 A–D). In frozen tissue sections of mice from three separate litters, T-cell and B-cell areas were disrupted, with no clear separation between the B-cell-rich lymphoid follicles and the T-cell-rich PALS (Fig. 3F). Clear segregation between the T-cell and B-cell areas in spleens from mice expressing low or no circulating LTβR-Fc protein was observed (Fig. 3E, and data not shown). The absence of the marginal zone surrounding the white pulp was most striking, especially seen at high magnification (Fig. 3D). Moreover, immunohistochemistry with antibodies specific for reticular fibroblasts (Fig. 4 A and B), marginal zone macrophages (Fig. 4 C and D), marginal zone metallophilic macrophages (Fig. 4 E and F), and MAdCAM-1 vascular addressin (MECA367: data not shown) confirmed the lack of the marginal zones. The absence of a reticular fibroblast network demonstrates that even the stromal elements of the marginal zone have not developed normally. However, the spleen was found to be phenotypically normal with respect to the percentage of cells expressing B- and T-cell surface antigens (data not shown).
Figure 3.
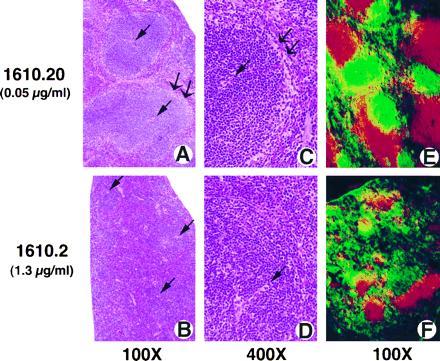
Blockade of LTαβ/LTβR interactions results in abnormal splenic architecture. Splenic tissue sections from mice expressing either low (A, C, and E) or high (B, D, and F) levels of the LTβR–Fc transgene, as described in Fig. 2. Formalin-fixed sections were stained with hematoxylin/eosin (A–D). A single arrow represents the location of the central arterioles, and a double arrow defines the marginal zones. Frozen tissue sections were stained with antibodies specific for T-cell (red) and B-cell (green) antigens (E and F), as described in Fig. 2. (A, B, E, and F, ×100; C and D, ×400.)
Figure 4.
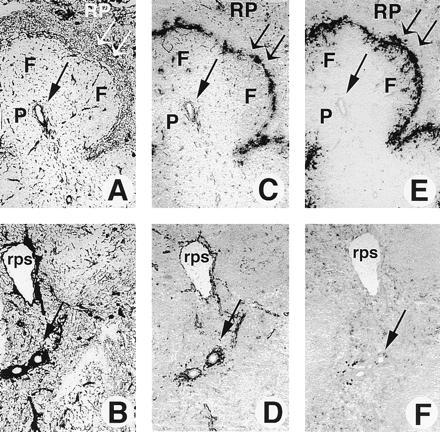
Absence of splenic marginal zones in mice expressing a soluble LTβR–Fc chimeric fusion protein. Frozen tissue sections of spleen from 10-week-old female mice expressing high levels of the LTβR–Fc protein (2.5 μg/ml, from founder 201) (B, D, and F) or transgenic-negative mice (A, C, and E) were stained with antibodies specific for reticular fibroblasts with ER-TR7 (A and B), marginal zone macrophages with ER-TR9 (C and D), or marginal zone metallophilic macrophages with MOMA-1 (E and F), as described. A single arrow represents the location of the central arterioles, and a double arrow defines the marginal zones. F, P, RP, and rps shows the location of the B-cell-rich follicles, the T-cell-rich PALS, the red pulp, and the red pulp sinus, respectively. (×125.)
DISCUSSION
LTα has clearly been shown to be required for peripheral lymphoid development (5, 6). Furthermore, it has been demonstrated that surface LTαβ interactions during days 11–16 of gestation are critical for normal lymph node development. Splenic T- and B-cell organization and Peyer’s patch development seem to require LTαβ interactions after day 17 of gestation (16). Consistent with this observation, our data demonstrate that high expression of the LTβR–Fc fusion protein after 3 days of life resulted in normal lymph node development; however, splenic and Peyer’s patch development were dramatically affected. Thus, this suggests that different secondary lymphoid organs require LTαβ/LTβR interactions at different times during development. Alternatively, different lymphoid organs may require distinct combinations of LTαβ- and LTβR-expressing lymphoid and stromal elements for organogenesis to occur.
The splenic marginal zone is composed of a network of reticular cells, predominantly containing marginal zone macrophages and resident IgM+, IgD− B lymphocytes, surrounding the white pulp, and separating it from the red pulp. Presumably, many activated LTαβ-expressing T and B cells pass through the marginal zone, trafficking to the PALS, and the follicles, respectively. The mechanism of this migration is unknown; however, it is believed that various types of marginal zone macrophages play a role (19). A specific dose-dependent effect of the LTβR–Fc chimeric protein was observed on splenic marginal zone development and T-cell/B-cell organization. Moreover, immunohistochemistry using monoclonal antibodies specific for marginal zone macrophages and reticular fibroblasts in conjunction with routine histology clearly demonstrated the absence of distinct marginal zones in spleens from mice expressing high levels of the LTβR–Fc fusion protein. Thus, blockade of LTαβ/LTβR interactions does not simply result in a down regulation of adhesion molecules by marginal zone cells, but results in the absence of the distinct populations of cells which make up the marginal zone. It is unclear if the lack of splenic marginal zones is directly responsible for the loss of splenic T-cell/B-cell organization in the mice expressing the LTβR–Fc chimeric protein. Alternatively, splenic stromal development and T-cell/B-cell organization may occur through mechanisms which are mutually exclusive.
Recently, a number of reports have suggested that both TNFRp60 and LTβR are able to signal crucial events required for normal lymphoid development and function (5–9, 16, 25, 26). It is becoming increasingly clear which receptor/ligand interactions are pivotal to these processes (Table 2). Although early reports demonstrated that LTα-deficient mice, but not TNFRp60- or p80-deficient mice, have a complete lack of lymph node development, closer evaluation has revealed that TNFRp60-deficient mice have splenic and Peyer’s patch abnormalities. Both TNFRp60- and LTα-deficient mice do not develop Peyer’s patches, have a lack of splenic marginal zone MAdCAM-1 expression, and do not form germinal centers following immunization (25, 26). In addition to a reduction of Peyer’s patches and an absence of splenic MAdCAM-1 expression, mice expressing a soluble LTβR–Fc fusion protein also lack splenic marginal zone MOMA-1, and ER-TR9 expression (Table 2). Lymph node development is also disrupted if surface LTαβ ligand is neutralized early in ontogeny (16). Expression of the LTβR–Fc transgene product did not appear to effect germinal center formation in Peyer’s patches. Germinal centers were present in all Peyer’s patches isolated from mice expressing the LTβR–Fc fusion protein. Germinal centers persist chronically in Peyer’s patches; however, in spleen and lymph nodes, germinal center formation follows antigen stimulation (27). The ability to form germinal centers in spleens of LTβR-Fc transgenic mice following antigen challenge is currently being evaluated.
Table 2.
Effects of TNF, LTα, and LTβ on lymphoid development
| Mice
|
|||
|---|---|---|---|
| LTα−/− | TNFRp60−/− | LTβR-Fc Tg | |
| Thymus | Normal | Normal | Normal |
| Lymph nodes | − | + | +* |
| Peyer’s patches | − | − | ↓ or − |
| Spleen | |||
| T-cell/B-cell areas | Disrupted | Normal | Disrupted |
| Marginal zones | − | + | − |
| MOMA-1 | − | + | − |
| ER-TR7 | ND | ND | − |
| ER-TR9 | ND | ND | − |
| MAdCAM-1 | − | − | − |
| Germinal centers | − | − | ND |
If absent, −; if present, +; ND, not determined.
no lymph node development if LTαβ is neutralized during days 11–16 of gestation (16).
Thus, if LTαβ only binds LTβR, and not other yet undetected receptor(s), then it appears that while TNFRp60 and LTβR are both essential for Peyer’s patch organogenesis, lymph node development requires only LTβR engagement (excluding mesenteric lymph nodes). The specific interactions necessary for splenic marginal zone formation, and T-cell/B-cell organization, also requires LTβR, although splenic MAdCAM-1 expression seems to be dependent on both TNFRp60 and LTβR engagement. Germinal center formation requires TNFRp60 and LTα; however, the requirement of surface LTαβ ligand in germinal center formation is still unclear. Blockade of, or a lack of, LTαβ/LTβR, TNF/TNFRp60 or LTα3/TNFRp60 interactions may also affect the expression of other receptor/ligand pairs required for these process to occur. Our results, together with the results of Rennert et al. (16) demonstrate that surface LTαβ (presumably signaling through the LTβR) is required for normal lymph node, splenic and Peyer’s patch development.
Acknowledgments
We thank C. M. Ambrose for providing the CA116 vector and the mutated hIgG1 construct; W. Force, C. Hession, and C. Ware for providing the LTβR cDNA which was used in the construction of the LTβR–Fc fusion protein; J. A. Nelson for discussion concerning human CMV expression; F. Mackay for the use of the mLTαβ bioassay in advance of publication as well as assistance in running the bioassays; R. Mebius for her valuable contribution to the immunohistological analysis; A. Cope, H.-K. Sytwu, and A. Marshak-Rothstein for critical discussion of the manuscript; and R. Pesich for technical assistance. This work was supported in part by grants from the Walter V. and Idun Y. Berry Foundation, the National Institutes of Health (Grant AI-36535, National Institute of Allergy and Infectious Diseases, and Grant CA-48734, National Cancer Institute), and the Department of Veterans Affairs.
Footnotes
Abbreviations: CMV, cytomegalovirus; LTα, lymphotoxin–α; LTαβ, lymphotoxin–αβ; LTβ, lymphotoxin–β; LTβR, lymphotoxin–β receptor; PALS, peri-arteriolar-lymphoid sheath; TNF, tumor necrosis factor; TNFRp60, TNF receptor-p60; TNFRp80, TNF receptor-p80.
References
- 1.Ware C F, VanArsdale T L, Crowe P D, Browning J L. Curr Top Microbiol Immunol. 1995;198:175–218. doi: 10.1007/978-3-642-79414-8_11. [DOI] [PubMed] [Google Scholar]
- 2.Tartaglia L A, Goeddel D V. Immunol Today. 1992;13:151–153. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- 3.Browning J L, Ngam-ek A, Lawton P, Demarinis J, Tizard R, Chow E P, Hession C, O’Brine-Greco B, Foley S F, Ware C F. Cell. 1993;72:847–856. doi: 10.1016/0092-8674(93)90574-a. [DOI] [PubMed] [Google Scholar]
- 4.Crowe P D, VanArsdale T L, Walter B N, Ware C F, Hession C, Ehrenfels B, Browning J L, Din W S, Goodwin R G, Smith C A. Science. 1994;264:707–710. [PubMed] [Google Scholar]
- 5.De Togni P, Goellner J, Ruddle N H, Streeter P R, Fick A, Mariathasan S, Smith S C, Carlson R, Shornick L P, Strauss-Schoenberger J, Russell J H, Karr R, Chaplin D D. Science. 1994;264:703–706. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 6.Banks T A, Rouse B T, Kerley M K, Blair P J, Godfrey V L, Kuklin N A, Bouley D M, Thomas J, Kanangat S, Mucenski M L. J Immunol. 1995;155:1685–1693. [PubMed] [Google Scholar]
- 7.Pfeffer K, Matsuyama T, Kundig T M, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi P S, Kronke M, Mak T W. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- 8.Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Nature (London) 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 9.Erickson S L, de Sauvage F J, Kikly K, Carver-Moore K, Pitts-Meek S, Gillett N, Sheehan K C F, Schreiber R D, Goeddel D V, Moore M W. Nature (London) 1994;372:560–563. doi: 10.1038/372560a0. [DOI] [PubMed] [Google Scholar]
- 10.Peppel K, Poltorak A, Melhado I, Jirik F, Beutler B. J Immunol. 1993;151:5699–5703. [PubMed] [Google Scholar]
- 11.Force W R, Walter B N, Hession C, Tizard R, Kozak C A, Browning J L, Ware C F. J Immunol. 1995;155:5280–5288. [PubMed] [Google Scholar]
- 12.Tao M-H, Smith R I F, Morrison S L. J Exp Med. 1993;178:661–667. doi: 10.1084/jem.178.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canfield S M, Morrison S L. J Exp Med. 1991;173:1483–1491. doi: 10.1084/jem.173.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lund J, Winter G, Jones P T, Pound J D, Tanaka T, Walker M R, Artymiuk P J, Arata Y, Burton D R, Jefferis R, Woof J M. J Immunol. 1991;147:2657–2662. [PubMed] [Google Scholar]
- 15.Miller G T, Hochman P S, Meier W, Tizard R, Bixler S A, Rosa M D, Wallner B P. J Exp Med. 1993;178:211–222. doi: 10.1084/jem.178.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rennert, D. P., Browning, J. L., Mebius, R. & Hochman, P. S. (1996) J. Exp. Med. 184, in press. [DOI] [PMC free article] [PubMed]
- 17.van Ewijk W, Ron Y, Monaco J, Kappler J, Marrack P, LeMeur M, Gerlinger P, Durand B, Benoist C, Mathis D. Cell. 1988;53:367–370. doi: 10.1016/0092-8674(88)90156-0. [DOI] [PubMed] [Google Scholar]
- 18.van Ewijk W. Annu Rev Immunol. 1991;9:591–615. doi: 10.1146/annurev.iy.09.040191.003111. [DOI] [PubMed] [Google Scholar]
- 19.Kraal G. Int Rev Cytol. 1992;132:31–73. doi: 10.1016/s0074-7696(08)62453-5. [DOI] [PubMed] [Google Scholar]
- 20.van Vliet E, Melis M, Foidart J M, van Ewijk W. J Histochem Cytochem. 1986;34:883–890. doi: 10.1177/34.7.3519751. [DOI] [PubMed] [Google Scholar]
- 21.Browning J L, Miatkowski K, Griffiths D A, Bourdon P R, Hession C, Ambrose C M, Meier W. J Biol Chem. 1996;271:8618–8626. doi: 10.1074/jbc.271.15.8618. [DOI] [PubMed] [Google Scholar]
- 22.Majeau G R, Meier W, Jimmo B, Kioussis D, Hochman P S. J Immunol. 1994;152:2753–2767. [PubMed] [Google Scholar]
- 23.Baskar J F, Smith P P, Nilaver G, Jupp R A, Hoffmann S, Peffer N J, Tenney D J, Colberg-Poley A M, Ghazal P, Nelson J A. J Virol. 1996;70:3207–3214. doi: 10.1128/jvi.70.5.3207-3214.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baskar J F, Smith P P, Ciment G S, Hoffmann S, Tucker C, Tenney D J, Coberg-Poley A M, Nelson J A, Ghazal P. J Virol. 1996;70:3215–3226. doi: 10.1128/jvi.70.5.3215-3226.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsumoto M, Mariathasan S, Nahm M H, Baranyay F, Peschon J J, Chaplin D D. Science. 1996;271:1289–1291. doi: 10.1126/science.271.5253.1289. [DOI] [PubMed] [Google Scholar]
- 26.Neumann B, Luz A, Peffer K, Holzmann B. J Exp Med. 1996;184:259–264. doi: 10.1084/jem.184.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griebel P J, Hein W R. Immunol Today. 1996;17:30– 39. doi: 10.1016/0167-5699(96)80566-4. [DOI] [PubMed] [Google Scholar]