

Abstract
Studies in melanoma patients have revealed that self proteins can function as targets for tumor-reactive cytotoxic T lymphocytes (CTL). One group of self proteins MAGE, BAGE, and GAGE are normally only expressed in testis and placenta, whilst another group of CTL recognized proteins are melanocyte-specific differentiation antigens. In this study we have investigated whether CTL can be raised against a ubiquitously expressed self protein, mdm-2, which is frequently overexpressed in tumors. The observation that T-cell tolerance is self major histocompatibility complex-restricted was exploited to generate CTL specific for an mdm-2 derived peptide presented by nonself major histocompatibility complex class I molecules. Thus, the allo-restricted T-cell repertoire of H-2d mice was used to isolate CTL specific for the mdm100 peptide presented by allogeneic H-2Kb class I molecules. In vitro, these CTL discriminated between transformed and normal cells, killing specifically Kb-positive melanoma and lymphoma tumors but not Kb-expressing dendritic cells. In vivo, the CTL showed antitumor activity and delayed the growth of melanoma as well as lymphoma tumors in H-2b recipient mice. These experiments show that it is possible to circumvent T-cell tolerance to ubiquitously expressed self antigens, and to target CTL responses against tumors expressing elevated levels of structurally unaltered proteins.
In the recent past a number of antigens recognized by cytotoxic T lymphocytes (CTL) isolated from melanoma patients have been identified at the molecular level. Although some melanoma-reactive CTL recognized novel antigens encoded by mutated genes, this was the exception and in the majority of cases the CTL recognized unaltered self proteins (1). The MAGE, BAGE, and GAGE proteins (2–4) are normally expressed only in the testis and the placenta, and aberrant expression in tumors can lead to the induction of CTL responses. The CTL can also recognize melanocyte lineage-specific antigens such as tyrosinase, Melan-A/Mart-1, gp100, and gp75 (5–9). There are several explanations why the CTL repertoire may not be tolerant to these proteins. First, they are not normally expressed in the thymus, and second, melanocytes may occupy immunologically privileged sites.
Mdm-2 is a zinc finger protein that can bind to p53 and neutralize its transcriptional activation properties (10). Mdm-2, p53 as well as the cell cycle control gene cyclin-D1 are frequently overexpressed in human tumors of different tissue origin (11–13), and therefore represent potential targets for tumor-reactive CTL. However, these self proteins are ubiquitously expressed in many different tissues including the thymus, where autoreactive CTL are likely to be deleted. In a recent study we found that the CTL repertoire of H-2b mice was indeed tolerant to the majority of peptides derived from murine mdm-2, cyclin-D1, and p53 (14). Only one peptide derived from mdm-2 failed to establish complete tolerance, and low numbers of potentially autoreactive CTL were detectable. This peptide bound inefficiently to H-2Db class I molecules that might explain why not all autoreactive CTL were deleted from the repertoire of H-2b mice. In contrast, tolerance to an mdm-2 peptide that bound strongly to H-2Kb class I molecules was complete, and high avidity CTL against this peptide were not detectable (14).
Here, we have investigated possibilities to overcome tolerance and to raise high avidity CTL against self peptides that bind efficiently to major histocompatibility complex (MHC) class I molecules. We took advantage of the observation that T lymphocyte tolerance is self MHC-restricted (15, 16). The CTL repertoire is rendered tolerant to peptides presented by self MHC class I molecules, but not to peptides presented by allogeneic class I alleles. Consequently, it may be possible to raise CTL specific for peptides presented by allogeneic class I molecules, even if the peptides are derived from normal cellular proteins. Such allo-restricted CTL may discriminate between physiological protein levels in normal cells and overexpression in tumors. To test these predictions we exploited the allo-restricted CTL repertoire of H-2d mice to raise CTL specific for an mdm-2 peptide presented by H-2Kb class I molecules.
METHODS
Animals.
C57BL/6 mice (H-2b) and BALB/c (H-2d) mice were supplied by the breeding colony of the Royal Postgraduate Medical School, Hammersmith Hospital (London). Mice were used at the age of 8–10 weeks.
Peptides.
The mdm100 peptide corresponds to amino acid 100–107 (YAMIYRNL) of the murine mdm-2 protein. In a previous study (14) we showed that this peptide bound efficiently to H-2Kb class I molecules. Other peptides derived from mdm-2 or cyclin-D1 that were found to bind to H-2Kb or Db class I molecules served as controls. All peptides used in this study were synthesized by the central peptide synthesis laboratory of the Imperial Cancer Research Fund (London), using fluorenylmethoxycarbonyl chemistry. The quality of the peptides was assessed by HPLC analysis and the expected molecular weight was observed using matrix-assisted laser desorption ionization mass spectrometry. The peptides were dissolved in PBS (pH 7.4) to give a concentration of 2 mM and stored at −20°C.
Cell Lines.
RMA cells (H-2b) originated from a Rauscher virus-induced C57BL/6N T-cell lymphoma. RMA-S cells were derived from RMA cells after mutagenesis with ethyl-methane-sulfonate followed by five rounds of selection with anti-H-2 alloantisera and rabbit complement treatment to obtain cells with decreased levels of MHC class I expression (17). RMA-S cells were found to have a point mutation at nucleotide 97 of the TAP2 gene, which generates a premature stop codon (18). B16-F1 and B16-F10 are variants of the C57BL/6 derived melanoma cell line B16, with low and high metastatic potential, respectively (19). The human cell line T2 is a fusion hybrid of a B-lymphoblastoid cell line and a T-lymphoblastoid cell. These cells have no TAP transporter genes and express reduced levels of HLA-A2 and no detectable endogenous HLA-B5 (20). T2 cells transfected with murine H-2Kb expressed Kb class I molecules at levels that were similar to the levels of HLA-A2. The T2-Kb cells were a gift from T. Elliott (John Radcliffe Hospital, Oxford).
CTL Induction.
Allo-restricted CTL were generated by in vitro stimulation of naive BALB/c splenocytes with peptide-coated RMA-S and T2-Kb stimulator cells. BALB/c splenocytes (4 × 106) were stimulated in 24-well plates with 4 × 105 RMA-S cells coated with mdm100 peptide in complete RPMI 1640 medium containing 10% fetal calf serum and 500 nM peptide. After 5 days, CTL were seeded in 96 well plates at 10, 100, and 1000 responder CTL per well. Each well contained 3 × 105 irradiated BALB/c splenocytes as feeders, and 104 irradiated T2-Kb stimulator cells that were previously pulsed with mdm100 peptides. The culture media was the same as above, except that recombinant interleukin 2 was added at a concentration of 10 units/ml. Fresh medium containing feeders and stimulator cells were added after 14 days, and after an additional five days each 96-well plate was tested in a CTL assay against mdm100-coated target cells and control targets. Microcultures that showed mdm100-specific killing were expanded and used for limiting dilution cloning on 96-well plates using 0.1, 1, and 10 CTL per well, 105 BALB/c splenocytes as feeders and 104 T2-Kb stimulator cells. CTL clones were expanded and used in the described experiments.
CTL Assays.
Cytotoxic activity was determined in 4-hr 51Cr-release assays against target cells coated with mdm100 peptides or MHC class I-binding control peptides as described (21). In some experiments dendritic cells were used as CTL targets. They were prepared from C57BL/10 splenocytes after removal of plastic adherent cells and centrifugation on a layer of 14.5% (wt/vol) metrizamide as described (22).
Isolation of Natural Peptides and HPLC Separation.
RMA lymphoma cells (4 × 108) were lysed in 4 ml of 2% trifluoracetic acid in H2O. The suspension was homogenized by ultrasonication and cell debris was removed by centrifugation in a centrifuge (Sigma 2K15) at ≈27,000 × g for 1 hr at 4°C. The supernatant was then transferred into Centricon 10 filter units (Amicon), which were spun at 5000 × g for 2.5 hr at 4°C. The filtrate containing peptides <10 kDa was HPLC separated over a Superpac PepS column (Pharmacia) using as buffer A 0.1% trifluoracetic acid in H2O and as buffer B 0.1% trifluoracetic acid in acetonitrile. The flow rate was 1 ml/min and the concentration of buffer B was increased from 0% to 60% at 1% per min. Fractions (1 ml) were collected, dried in a Servant speed vacuum drier, and resuspended in 100 μl PBS. A total of 10 μl of each HPLC fraction was used to coat 51Cr-labeled T2-Kb cells, which then served as targets for mdm100 peptide-specific CTL.
RESULTS
Isolation of High Avidity CTL from the Allo-Restricted Repertoire.
In a previous study we identified an mdm-2-derived peptide (mdm100) that efficiently bound to H-2Kb class I molecules (14). However, in C57BL/6 mice (H-2b) this peptide only stimulated low avidity CTL that recognized target cells coated with the synthetic mdm100 peptide, but not target cells expressing mdm-2 endogenously. One possible explanation was that high avidity CTL that can recognize low concentrations of naturally produced peptides were deleted from the repertoire of H-2b mice. Since tolerance is MHC-restricted, the CTL repertoire of BALB/c mice (H-2d) would not be expected to be tolerant to mdm100 peptides presented by H-2Kb class I molecules, and it should be possible to isolate high avidity CTL. Thus, allo-restricted CTL were generated by stimulating naive BALB/c splenocytes with peptide-coated RMA-S (H-2b) and T2-Kb cells. Thirty-three CD8+ CTL clones were isolated that lysed RMA-S and T2-Kb cells only when they were coated with mdm100 peptides, but not when they were coated with other H-2b class I binding peptides (Fig. 1 B and C). Syngeneic P815 cells (H-2d) were unable to present the mdm100 peptide to these H-2d-derived CTL clones (data not shown). Peptide titration experiments revealed that 16 clones were of high “avidity” requiring 10−12 to 10−14 molar peptide concentration for half maximal target cell lysis (Fig. 1A). In contrast, 17 CTL clones were of low “avidity” requiring 10−8 to 10−9 molar peptide concentration (Fig. 1A).
Figure 1.
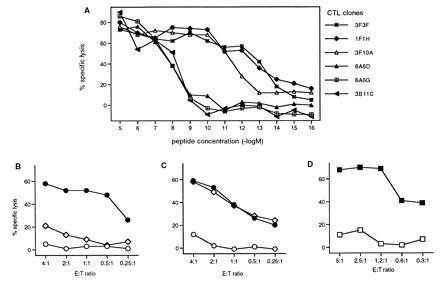
Characterization of allo-restricted CTL clones specific for the mdm100 peptide. (A) Recognition of peptide-coated T2-Kb cells by six CTL clones specific for the mdm100 peptide. In total, 33 CTL clones were analyzed and the peptide titration curves for 16 were similar to that of the clones 3F3F, 1F1H and 3F10A, while 17 clones showed titration curves similar to that of the clones 3B11C, 6A6G, and 6A6D. B and C show lysis of RMA cells (◊) and RMA-S cells coated with either mdm100 peptides (•) or with class I binding control peptides (○) by a representative low-avidity CTL clone 6A6G (B) or by a representative high-avidity clone 3F3F (C). (D) Lysis of dendritic cells coated with either mdm100 peptides (▪) or with class I binding control peptides (□) by high-avidity clone 3F11A.
Allo-Restricted CTL Lyse Tumor Cells but Not Normal Cells.
The avidity of CTL clones was functionally important, since it determined the level of tumor cell lysis. All high-avidity CTL clones efficiently killed RMA lymphoma cells (H-2b), whilst lysis by all low avidity CTL clones was inefficient (Fig. 1 B and C). To determine whether high-avidity CTL could discriminate between transformed and normal cells, we used dendritic cells as targets because they express high levels of MHC class I molecules as well as costimulatory molecules that are important for T-cell activation (23). The mdm-2 expression levels in dendritic cells have not yet been determined, but they might be similar to those found in tissues that consist primarily of nonproliferating cells. For example brain, heart and muscle contained considerable levels of mdm-2 RNA (24), although the levels were higher in tissues with a high proliferation index such as testis and thymus. Fig. 1D shows that the levels of mdm-2 expression in dendritic cells were insufficient to trigger lysis by high-avidity CTL clones. Importantly, dendritic cells coated with the mdm100 peptide were efficiently lysed, indicating that these cells were not inherently resistant to CTL mediated killing (Fig. 1D). These in vitro experiments indicated that high-avidity CTL could discriminate between transformed RMA tumor cells and normal dendritic cells. In vivo experiments also suggested that normal tissues were not attacked by high-avidity CTL, since intravenous injection of 107 CTL on three consecutive days was well tolerated by H-2Kb-positive C57BL/6 mice (data not shown).
Tumor cell recognition by high-avidity CTL was not limited to RMA lymphoma cells, but was also seen against B16 melanoma cells. However, melanoma recognition was dependent upon the levels of MHC class I expression. CTL killing was observed against the Kb-positive B16-F10 melanoma variant (Fig. 2B), whilst the Kb-negative B16-F1 variant was resistant to CTL lysis (Fig. 2A). The relative inefficient lysis of B16-F10 (Fig. 2B) compared with RMA (Fig. 1C) correlated with low and high levels of Kb expression in these tumor cells (data not shown). Increasing the density of Kb/peptide ligands by coating B16-F10 cells with mdm100 peptides resulted in enhanced CTL killing (Fig. 2B).
Figure 2.
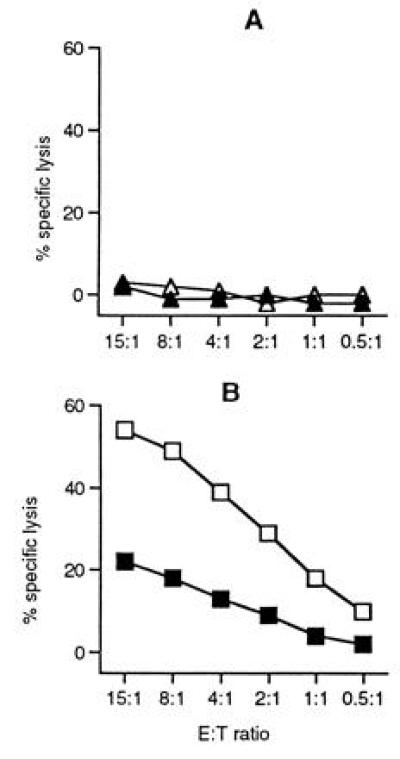
Recognition of B16 melanoma cells by high-“avidity” mdm100-specific CTL. A representative high-avidity CTL clone (1F7E) was analyzed in a 4-hr 51Cr-release assay against the nonmetastatic B16-F1 melanoma variant (A) and against the metastatic variant B16-F10 (B). B16-F1 and B16-F10 cells were either coated with mdm100 peptides (open symbols) or with MHC class I binding control peptides (solid symbols). Staining experiments revealed that B16-F1 cells were H-2Kb negative and that B16-F10 cells expressed low levels H-2Kb (not shown).
Evidence That the mdm100 Peptide Is Naturally Produced in Tumor Cells.
The experiments described above have shown that CTL clones raised against the synthetic mdm100 peptide were able to recognize Kb-positive tumor cells. To address whether fortuitous cross-recognition of unrelated peptides accounted for CTL recognition of tumor cells, low molecular weight peptides were isolated from RMA cells and separated by reverse phase HPLC. Individual HPLC fractions were collected and used to coat T2-Kb cells, which were then used as CTL targets. Only fractions 31 and 29 contained CTL recognized peptides (Fig. 3B). HPLC separation of synthetic mdm100 peptides revealed a major peptide peak in fraction 31 and minor peaks in fraction 29 (Fig. 3A). Mass spectrometry revealed that fraction 31 contained the mdm100 peptide (YAMIYRNL) and fraction 29 contained the same peptide with an oxidized methionine at position 3. The coelution of synthetic and naturally produced peptides strongly suggest that RMA tumor cells naturally produce the mdm100 peptide. The oxidized version of this peptide is probably not naturally produced, but might be the product of methionine oxidation during the trifluoracetic acid isolation procedure. Formal proof that RMA cells produce the mdm100 peptide would require sequence analysis by Edman degradation or mass spectrometry of material present in fractions 29 and 31 (Fig. 3B).
Figure 3.
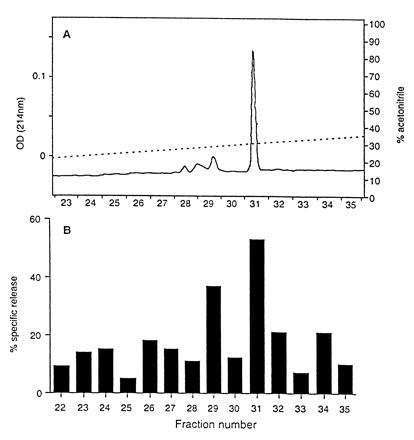
CTL recognition of naturally produced peptides extracted from RMA lymphoma cells. (A) HPLC elution profile of 1 μg of the synthetic peptide mdm100. (B) CTL recognition profile of HPLC fractions containing naturally produced peptides extracted from RMA cells. Peptides were prepared from RMA lymphoma cells as described and separated by HPLC using the same conditions as in A.
Allo-Restricted CTL Can Delay Tumor Growth in H-2b Mice.
High-avidity CTL were tested for their ability to control the growth of RMA lymphoma and B16-F10 melanoma tumors in vivo. C57BL/6 mice injected with RMA cells developed tumors at day 9 and died of large tumor burden within 14–15 days (Fig. 4 A and B). In contrast, tumor development was completely inhibited for 15–17 days in mice that were injected with RMA cells and CTL. The level of tumor protection was dose dependent and greater in mice injected with a large dose of CTL (Fig. 4A), compared with mice injected with a low dose of CTL (Fig. 4B). Similar results were obtained with the B16-F10 melanoma. Mice injected with B16-F10 cells only developed tumors after eight days and died of tumor burden after 11–13 days, whilst mice injected with B16-F10 cells and CTL were tumor free until day 15 (Fig. 4C). Immune responses of recipient H-2b mice against the injected H-2d-derived CTL clones is expected to limit their half life in vivo, and may explain why tumor protection was not complete. Further experiments will reveal to what extent immunosuppression of recipient mice can enhance tumor protection by transferred CTL clones. The experiments described here have established that allo-restricted CTL clones specific for peptides derived from the normal mdm-2 protein can potentially control the growth of lymphoma and melanoma tumors in vivo.
Figure 4.
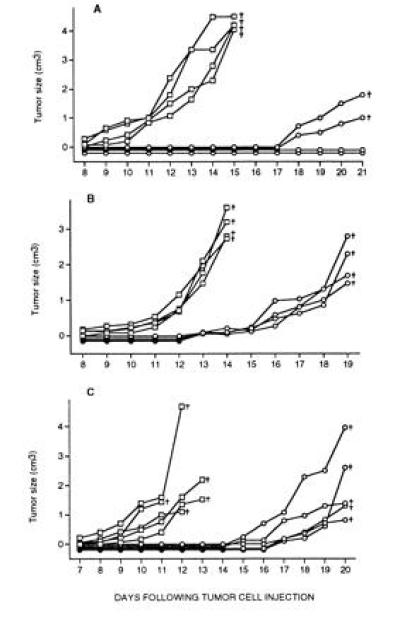
Control of tumor growth by high-avidity, mdm100-specific CTL. (A) Eight C57BL/10 mice were injected subcutaneously with 105 RMA lymphoma cells (□) or with 105 lymphoma cells together with 106 CTL (○). (B) Eight C57BL/10 mice were similarly injected with 5 × 105 RMA lymphoma cells (□) or with 5 × 105 lymphoma cells together with 5 × 105 CTL (○). (C) Ten C57BL/6 mice were injected with either 105 B16-F10 melanoma cells (□), or with 105 B16-F10 cells together with 106 CTL (○). Similar results were obtained when either individual high-avidity CTL clones or a mixture of clones were used for these experiments. Mice were monitored daily and the tumor growth for each individual mouse is shown. The crosses indicate that the mice died or were sacrificed because of large tumor burden.
DISCUSSION
We have demonstrated that it is possible to circumvent self tolerance by exploiting specificities present in the allo-restricted CTL repertoire. CTL clones specific for a peptide presented by nonself H-2Kb class I molecules were obtained from the allo-restricted repertoire of BALB/c mice. Approximately 50% of the CTL clones were of high avidity and efficiently recognized mdm-2 expressing tumor cells, whilest low avidity CTL only recognized targets coated with synthetic peptides. The high-avidity CTL clones displayed tumor-specificity and did not lyse dendritic cells or concanavalin A-stimulated T-cell blast in vitro (not shown). Although intravenous injection of high-avidity CTL clones was well tolerated by H-2b recipient mice, further experiments are required to exclude the possibility that some normal tissues might be recognized by these CTL clones.
In a previous study we found that the repertoire of H-2b mice was deleted of high-avidity CTL specific for the mdm100 peptide. This peptide stimulated only low-avidity CTL that did not recognize mdm-2 expressing tumor cells (14). Therefore, the isolation of CTL capable of recognizing tumor cells was constrained by tolerance to the mdm-2 protein. Similarly, targeting of tumors overexpressing normal p53 protein was also limited by CTL tolerance. In an elegant series of experiments (25), the CTL repertoire of HLA-A2.1 transgenic mice was exploited to circumvent tolerance to human p53. The CTL repertoire of these transgenic mice was tolerant only to murine p53 peptides. Two p53 peptides with sequence differences between man and mouse were found to stimulate high-avidity CTL that recognized HLA-A2.1-positive tumor cells but not dendritic cells. These studies showed that HLA transgenic mice can be exploited to generate murine CTL against human tumors overexpressing cellular proteins. This approach is dependent upon the availability of transgenic mice, and is limited to peptides with sequence differences between man and mouse. The advantage of the approach described here is that CTL can be generated against any peptide expressed at elevated levels in tumors.
Can allo-restricted CTL clones be exploited for adoptive tumor immunotherapy? Immune responses by immunocompetent recipients against transferred, allogeneic CTL clones might be a major limitation. Therefore, adoptive immunotherapy with these CTL clones may be most effective in immunosuppressed cancer patients. For example, patients with chronic myeloid leukemia are ideally suited for immunotherapy with allo-restricted CTL clones. chronic myeloid leukemia patients frequently receive allogeneic bone marrow transplants, which requires immunosuppression to favor bone marrow engraftment. It has been known for some time that the prognosis of these patients is improved when donor derived T lymphocytes mount an immune response against patient’s leukemic cells (26). However, this graft-versus-leukemia reaction is often associated with detrimental graft-versus-host disease. The murine experiments described here indicate that it is possible to generate allo-restricted CTL clones that can specifically kill leukemic cells. These results suggest that it should be possible isolate human CTL clones specific for peptides presented at elevated levels in leukemic cells. Such CTL clones could be used for adoptive immunotherapy of chronic myeloid leukemia patients, and would be expected to mediate antileukemia effects without causing graft-versus-host disease.
Acknowledgments
We thank Drs. E. Simpson, D. Gray, and R. Lechler for critically reading the manuscript. We thank Dr. I. Hart for providing the B16-F1 and B16F-10 cells. We are also grateful to Nicola O’Reilly and Elizabeth Li who synthesized the peptides used in this study. This work was supported by the Cancer Research Campaign.
Footnotes
Abbreviations: CTL, cytotoxic T lymphocytes; MHC, major histocompatibility complex.
References
- 1.Boon T, van der Bruggen P. J Exp Med. 1996;183:725–729. doi: 10.1084/jem.183.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Bruggen P, Traversari C, Chomez P, Lurquin C, de Plaen E, van den Eynde B, Knuth A, Boon T. Science. 1991;254:1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 3.Boel P, Wildmann C, Sensi M L, Brasseur R, Renauld J C, Coulie P, Boon T, van der Bruggen P. Immunity. 1995;2:167–75. doi: 10.1016/s1074-7613(95)80053-0. issn, 1074–7613. [DOI] [PubMed] [Google Scholar]
- 4.Van den Eynde B, Peeters O, De Backer O, Gaugler B, Lucas S, Boon T. J Exp Med. 1995;182:689–98. doi: 10.1084/jem.182.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brichard V, Van Pel A, Wolfel T, Wolfel C, De Plaen E, Lethe B, Coulie P, Boon T. J Exp Med. 1993;178:489–495. doi: 10.1084/jem.178.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coulie P G, Brichard V, Van Pel A, Wolfel T, Schneider J, Traversari C, Mattei S, De Plaen E, Lurquin C, Szikora J P, Renauld J C, Boon T. J Exp Med. 1994;180:35–42. doi: 10.1084/jem.180.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawakami Y, Eliyahu S, Delgado C H, Robbins P F, Rivoltini L, Topalian S L, Miki T, Rosenberg S A. Proc Natl Acad Sci USA. 1994;91:3515–3519. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bakker A B H, Schreurs M W J, de Boer A J, Kawakami Y, Rosenberg S A, Adema G J, Figdor C G. J Exp Med. 1994;179:1005–1009. doi: 10.1084/jem.179.3.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cox A L, Skipper J, Chen Y, Henderson R A, Darrow T L, Shabanowitz J, Engelhard V H, Hunt D F, Slingluff C L., Jr Science. 1994;264:716–719. doi: 10.1126/science.7513441. [DOI] [PubMed] [Google Scholar]
- 10.Wu X, Bayle J H, Olson D, Levine A J. Genes Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 11.Bueso Ramos C E, Yang Y, deLeon E, McCown P, Stass S A, Albitar M. Blood. 1993;82:2617–2623. [PubMed] [Google Scholar]
- 12.Bartkova J, Lukas J, Muller H, Lutzhoft D, Strauss M, Bartek J. Int J Cancer. 1994;57:353–361. doi: 10.1002/ijc.2910570311. [DOI] [PubMed] [Google Scholar]
- 13.Bartek J, Bartkova J, Vojtesek B, Staskova Z, Lukas J, Rejthar A, Kovarik J, Midgley C A, Gannon J V, Lane D P. Oncogene. 1991;6:1699–1703. [PubMed] [Google Scholar]
- 14.Dahl A M, Beverley P, Stauss H J. J Immunol. 1996;157:239–246. [PubMed] [Google Scholar]
- 15.Matzinger P, Zamoyska R, Waldmann H. Nature (London) 1984;308:738–741. doi: 10.1038/308738a0. [DOI] [PubMed] [Google Scholar]
- 16.Rammensee H G, Bevan M J. Nature (London) 1984;308:741–744. doi: 10.1038/308741a0. [DOI] [PubMed] [Google Scholar]
- 17.Ljunggren H-G, Kärre K. J Exp Med. 1985;162:1745–1759. doi: 10.1084/jem.162.6.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y, Früh K, Chambers J, Waters J B, Wu L, Spies T, Peterson P A. J Immunol. 1992;267:11669–11672. [PubMed] [Google Scholar]
- 19.Fidler I J, Nicolson G L. J Natl Cancer Inst. 1976;57:1199–1202. doi: 10.1093/jnci/57.5.1199. [DOI] [PubMed] [Google Scholar]
- 20.Alexander J, Payne J A, Murray R, Frelinger J A, Cresswell P. Immunogenetics. 1989;29:380–388. doi: 10.1007/BF00375866. [DOI] [PubMed] [Google Scholar]
- 21.Sadovnikova E, Zhu X, Collins S, Zhou J, Vousden K, Crawford L, Beverley P, Stauss H J. Int Immunol. 1993;6:289–296. doi: 10.1093/intimm/6.2.289. [DOI] [PubMed] [Google Scholar]
- 22.Macatonia S E, Taylor P M, Knight S E, Askonas B A. J Exp Med. 1989;169:1255–1264. doi: 10.1084/jem.169.4.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steinman R M. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 24.Fakharzadeh S S, Trusko S P, George D L. EMBO J. 1991;10:1565–1569. doi: 10.1002/j.1460-2075.1991.tb07676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Theobald M, Biggs J, Dittmer D, Levine A J, Sherman L A. Proc Natl Acad Sci USA. 1995;92:11993–11997. doi: 10.1073/pnas.92.26.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldman J M. Bone Marrow Transplant. 1989;1:133–134. [PubMed] [Google Scholar]