

Abstract
Epigenetic chromatin remodeling and modifications of DNA represent central mechanisms for regulation of gene expression during brain development and in memory formation. Emerging evidence implicates epigenetic modifications in disorders of synaptic plasticity and cognition. This review focuses on recent findings that HDAC inhibitors can ameliorate deficits in synaptic plasticity, cognition and stress-related behaviors in a wide range of neurologic and psychiatric disorders including Huntington’s disease, Parkinson’s disease, anxiety and mood disorders, Rubinstein-Taybi syndrome and Rett syndrome. These agents may prove useful in the clinic for the treatment of the cognitive impairments that are central elements of many neurodevelopmental, neurological and psychiatric disorders.
Introduction
Recent findings implicate epigenetic modifications in the etiology of neurodegenerative and psychiatric disorders. Covalent epigenetic modifications of conserved lysine residues in the amino-terminal tails of histone proteins and methylation of DNA at CpG dinucleotides control accessibility of chromatin to the core transcriptional machinery and play an essential role in determining the activation state of genes. An array of post-translational histone modifications are known and these include acetylation, methylation, ubiquitination, phosphorylation and SUMOlyation, all of which can serve as epigenetic tags (for review, see [1–3]). These modifications alter chromatin structure and make specific regions of the genome more or less accessible to the transcriptional machinery. Epigenetic remodeling is crucial to cellular differentiation, development and behavior, including learning and memory [1,4,5]. The epigenome is responsive to synaptic activity and provides a link between experience, genetic predisposition, and changes in neural function. Chromatin modifications are not static but dynamically change in a cell-specific manner during development and in response to external stimuli including neuronal insults. Epigenetic dysregulation is a common theme in disorders of synaptic plasticity and cognition including neurodegenerative disorders (Huntington’s disease, Parkinson’s disease and ischemia), mood disorders (depression and anxiety) and neurodevelopmental disorders (Rubinstein-Taybi syndrome, Rett syndrome and Fragile X syndrome). This article reviews new insight into the molecular mechanisms underlying activity-dependent remodeling of the epigenome under physiological and pathological conditions and highlights the importance of epigenetic modifications in developing new therapeutic approaches for psychiatric and neurological disorders.
Histone modifications and transcriptional regulation
Histone modifications regulate gene expression in three mechanistic ways [1–3]. First, they regulate chromatin structure, making genetic loci more or less accessible to the transcriptional machinery. Second, they serve a signaling role by integrating responses to multiple biochemical signaling cascades and recruit or repel the transcriptional machinery and chromatin remodeling complexes. Third, and perhaps most interestingly, histone modifications mediate epigenetic changes in gene expression that provide a mechanism by which neural function and behavior are stably altered for long periods of time in response to transient experience or neuronal insult. An emerging concept is that acetylated and methylated histone residues function as a complex epigenetic surface that is interpreted by a rapidly expanding family of proteins that are epigenetic “readers” [2,6]. There is also an interaction between histone modifications and the methylation of CpG dinucleotides at distinct genomic locations, and DNA methylation can “cement” changes in gene expression initially driven by histone modifications. Methylated DNA is read by proteins like methyl-CpG binding protein 2 (MeCP2), a protein that functionally links DNA methylation to gene silencing [1–3].
Histone acetyltransferases
Histone acetylation has received much attention in the nervous system in part because of our knowledge of the enzymatic machinery and signal transduction mechanisms that regulate this post-translational modification. Acetylation of core histones is catalyzed by transcriptional coactivators such as CREB-binding protein (CBP), which possess histone acetyltransferase (HAT) activity [1,6]. Histone acetylation remodels chromatin structure, thereby modulating transcription [1,6]. Specificity of gene regulation is achieved by the recruitment of HATs by transcription factors to specific genetic loci, where they locally modify histones. Importantly, HATs interact with a large number of transcription factors and thus serve as critical hubs, integrating the activity of multiple signaling cascades. Acetylation is rapidly and reversibly induced in an experience-dependent manner, but can be long-lived. HATs are evolutionarily conserved from yeast to humans and are categorized in three main families, GNAT, MYST and CBP/p300, according to the structure of the catalytic domains [1,6]. In addition to their catalytic domains, HATs and other chromatin-associated proteins contain bromodomains, evolutionarily conserved domains that bind to acetylated residues [6]. Acetylated histones serve as epigenetic markers or ‘tags’, which recruit HATs and other bromodomain-containing proteins.
Histone deactylases
Histone deactylases (HDACs) remove acetyl groups from lysine/arginine residues in the amino-terminal tails of core histones and other proteins, thus reversing the effects of the HATs [1,6]. Deacetylation of histone proteins shifts the balance towards chromatin condensation and thereby silences gene expression. Unlike HATs, HDACs have a rich structural diversity, which confers diversity of function and renders HDACs promising targets for drug discovery and therapeutic intervention. Many HDACs share a common zinc-dependent catalytic domain but differ in accessory domains that confer specificity of targets and regulation. Altogether, mammalian HDACs fall into four main classes, Class 1–4, with class 1 and class 2 HDACs receiving the most attention in the nervous system [7]. Class 1 HDACs (HDACs 1, 2, 3 and 8) are homologs of the yeast HDAC RPD3, are constitutively nuclear proteins and are widely expressed. Class 2 HDACs (HDACs 4, 5, 6, 7, 9 and 10) are homologs of yeast Hda1, are expressed in a tissue- and cell-specific manner and are regulated, at least in part, by shuttling between the nucleus and cytoplasm, where they are stabilized by interactions with 14-3-3 proteins. Sequence specificity of HDAC action is acquired by recruitment of HDACs to specific genetic loci by repressors, co-repressors and methyl-DNA binding proteins.
HDAC inhibitors
Recent years have witnessed an explosion in the development of new HDAC inhibitors [7]. HDAC inhibitors can be classified into four main chemical families, the short-chain fatty acids (e.g., sodium butyrate, phenylbutyrate and valproic acid), the hyroxamic acids (e.g., trichostatin A and suberoylanilide hydroxamic acid (SAHA)), the epoxyketones (e.g., trapoxin) and the benzamides. Of these, the most widely studied are sodium butyrate, phenylbutyrate, trichostatin A and SAHA. The butyrates are known to cross the blood-brain barrier [7,8]. The initial interest in these inhibitors came from studies linking HDACs to a wide variety of human cancers. HDAC inhibitors arrest growth, induce differentiation and, in some cases, apoptosis and have potent anit-cancer activities, with remarkable tumor specificity [7,8]. For this reason, inhibitors of class 1 and 2 HDACs are in phase I/II clinical trials for cancer therapy and potentially cancer prevention. In the nervous system, the anticonvulsant and mood-stabilizing drug valproic acid was identified as an inhibitor of HDAC1, thereby linking its anti-epileptic effects to changes in histone acetylation. As described below, more recent work has revealed that inhibitors of class 1 and 2 HDACs represent novel therapeutic approaches to treat neurodegenerative disorders, depression and anxiety and the cognitive deficits that accompany many neurodevelopmental disorders.
HDAC inhibitors as potential therapeutics for neurodegenerative disorders
Huntington’s disease
A role for HDACs in neurodegenerative disorders was first hinted at by experiments that showed that HDAC inhibitors ameliorate the cognitive and motor deficits characteristic of Huntington’s disease (HD). HD is an inherited, late onset autosomal-dominant neurodegenerative disorder characterized by progressive motor, psychiatric and cognitive decline that affects one person per 10,000 people of Western European descent [9]. Marked neuronal loss occurs in the cortex and striatum. HD is caused by a polyglutamine (CAG) expansion in the 5’ coding region of the huntingtin (htt) gene [9,10]. Transcriptional dysregulation is central to the pathogenesis of HD. Mutant huntingtin localizes primarily to the nucleus where it forms aggregates of mutant polyQ protein, which bind and functionally impair transcription factors and coactivators such as CBP [9,10]. Loss of accessible CBP leads to dysregulation of CBP/CREB-mediated gene expression, histone deacetylation and ultimately, neuronal death. Early studies demonstrated the ability of HDAC inhibitors to rescue lethality and photoreceptor neurodegeneration in a Drosophila model of polyglutamine disease [11]. These findings were extended to the mouse models of HD by several laboratories, who showed that treatment with HDAC inhibitors such as sodium butyrate and phenylbutyrate attenuate neuronal loss, increase motor function and extend survival in R6/2 mice [12–14]. These findings strengthen the idea that epigenetic dysregulation plays a critical role in the pathogenesis of HD and suggest that therapeutics that correct alterations in epigenetic modifications may be efficacious even after the onset of the disease process.
Parkinson’s disease
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that affects 1% of the population over 65 [15]. Approximately six million people worldwide have PD. To date, no treatment has been identified that halts the death of dopaminergic neurons that underlies PD. Although the etiology of PD is not well understood, familial PD can arise as a consequence of deficiency in the neuronal protein α-synuclein. Recent studies indicate that HDAC inhibitors may represent a promising therapeutic strategy to ameliorate the progressive neurodegeneration associated with PD. The initial link between PD and epigenetic dysregulation came from studies of Drosophila. Using a Drosophila model of PD, Feany and colleagues demonstrated that nuclear targeting of α-synuclein promotes its toxicity and that sequestration of α-synuclein to the cytoplasm is protective [16]. It was further shown that α-synuclein binds directly to histones, reduces levels of acetylated histone H3 and inhibits HAT-mediated acetyltransferase activity [16]. These findings implicate α-synuclein in the degeneration associated with PD. Administration of HDAC inhibitors in vivo or in vitro rescued α-synuclein- induced toxicity [16]. These findings underscore the potential of HDAC inhibitors for therapeutic intervention in the neurodegeneration and cognitive impairments in PD. Future studies to examine the efficacy of HDAC inhibitors in mouse models of PD are warranted.
Alzheimer’s disease
Another progressive neurodegenerative disorder characterized by aberrant CREB/CBP signaling is Alzheimer’s disease (AD). AD is a neurodegenerative disorder that currently affects nearly 2% of the population in industrialized countries; the risk of AD dramatically increases in individuals beyond the age of 70 and it is predicted that the incidence of AD will increase threefold within the next 50 years (http://www.alz.org). Hallmark neuropathological features of AD include extracellular plaques and intracellular tangles [17]. Endoproteolytic cleavage of the transmembrane amyloid precursor protein (APP) generates β-amyloid peptides, which aggregate to form plaques. A common theme in neurodegenerative disorders (HD, PD and AD) is the concept that intraneuronal aggregates such as plaques interfere with transcription and cause deficits in plasticity and cognition [17].
Although not as clearly defined in AD as in other neurodegenerative disorders, intriguing findings of Sudhof and colleagues suggest a possible role for transcriptional dysregulation in AD. The α- and β-secretases initially cleave APP at defined extracellular sequences. Subsequent cleavage of APP by γ-secretase generates not only the extracellular fragment β-amyloid, but also an intracellular tail fragment. The tail fragment recruits the nuclear adaptor protein Fe65 and the histone acetyltransferase Tip60; this complex potently stimulates transcription, suggesting that release of the cytoplasmic tail of APP by γ-cleavage may function in gene expression [18–20]. These findings suggest the intriguing notion that the neuropathology associated with AD is due, at least in part, to epigenetic dysregulation.
Although no direct demonstration of amelioration of synaptic plasticity or cognitive impairments have been documented for AD, recent studies from the laboratory of Tsai and colleagues show that HDAC inhibitors restore histone acetylation status and learning and memory in a mouse model of neurodegeneration [21]. This group conditionally and inducibly expressed p25, a cell-cycle protein implicated in neurodegenerative disease, under the control of the CaMKII promoter. The effect of HDAC inhibitors could be mimicked by exposure of the mice to a combination of complex inanimate and social stimulation (environmental enrichment), consistent with the notion that experience-dependent plasticity acts via epigenetic remodeling to ameliorate plasticity and cognitive deficits.
Ischemic stroke
Stroke is the third leading cause of death in the United States and the primary cause of disabilities in adults [22]. Of the 600,000 new victims each year, 30% die and another nearly 30% become severely and permanently disabled. Most Stage III clinical trials conducted to date have failed. It is thus critical that new molecular mechanistic information be revealed and be brought into the translational arena with the goal of developing novel treatment strategies for neuroprotection, rescue and repair in order to ameliorate the deleterious consequences of stroke. Global ischemia can arise as a consequence of cerebrovascular accidents, cardiac arrest, cardiac surgery, profuse bleeding, near-drowning and carbon monoxide poisoning. Brief ischemic insults cause selective, delayed death of hippocampal CA1 neurons and severe cognitive deficits. The substantial delay between insult and cell death is consistent with a role for transcriptional changes.
The transcriptional repressor RE1 silencing transcription factor (REST, also called NRSF) is widely expressed during embryogenesis and plays a critical role in terminal neuronal differentiation [23–25]. In neural progenitors and non-neural cells, REST actively represses a large array of neural-specific genes important to synaptic plasticity including synaptic vesicle proteins, structural proteins, voltage-gated channels and ligand-gated receptors, allowing non-neuronal transcripts to be expressed [23–25]. As neurons differentiate, REST orchestrates a set of epigenetic modifications that distinguish neuronal from non-neuronal cells. In global ischemia, selectively vulnerable hippocampal neurons exhibit aberrant accumulation of REST/NRSF in the nucleus and REST-dependent silencing of target genes essential for neuronal function [26]. A fundamental mechanism by which REST silences target genes is by recruitment of MeCP2 and corepressor complexes that promote histone H3 lysine 9 (H3K9) deacetylation and methylation. Recent findings that REST recruits CoREST, G9a and MeCP2 to promoters of target genes and promotes epigenetic remodeling of target genes in selectively vulnerable hippocampal neurons implicates REST-dependent epigenetic remodeling in the pathogenesis of global ischemia [27]. Dysregulation of REST and its target genes is implicated not only in global ischemia, but also in the pathogenesis of Down syndrome, Alzheimer’s disease, Huntington’s disease, epilepsy and X-linked mental retardation [23–25].
Two recent studies demonstrate a role for HDAC inhibitors in amelioration of neuronal death and cognitive deficits in postischemic neurons. In one study, Chiarugi and colleagues report that the potent HDAC inhibitor SAHA administered intraperitoneally to mice at 0 and 6 h after induction of ischemic stroke by middle cerebral artery occlusion (MCAO) prevented H3 deacetylation, promoted expression of neuroprotective proteins Bcl-2 and Hsp70 and reduced infarct volume, indicating a neuroprotective action for SAHA [28]. In a second study, Moskowitz, Dirnagl and colleagues observed aberrant DNA methylation in the brains of wild-type mice subjected to mild ischemic brain injury by the MCAO model; administration of the demethylating agent 5-aza-2’-deoxycytidine and the HDAC inhibitor trichostatin A conferred stroke protection in wild-type mice subjected to mild, but not severe ischemic damage [29]. These findings underscore the therapeutic potential of HDAC inhibitors for therapeutic intervention in the neurodegeneration associated with stroke.
HDAC inhibitors: Potential therapeutics for mood and anxiety disorders
Depression
One psychiatric disorder for which strong evidence exists to suggest that histone acetylation represents a valid therapeutic target is depression [30]. Major depressive disorder is a very common disorder, with a lifetime risk of 16.2% in the United States [31]. Thus, the development of novel antidepressants with greater efficacy would serve a large potential patient market. One drawback of current antidepressant medications, most of which act to increase synaptic levels of serotonin or norepinephrine, is the slow onset of action, in many cases on the order of 2–4 weeks. This delay between the administration of an antidepressant and the appearance of therapeutic effects suggests that the elevated synaptic levels of serotonin and norepinephrine must alter other processes that then lead to therapeutic effects. Several target processes have been identified, but a common thread is that elevated levels of neurotransmitters may alter the expression of specific target genes, such as BDNF, whose products act to alter neural function and behavior. Developing therapeutics to target processes downstream of neurotransmitter action is a promising avenue of research. Work from the laboratory of Eric Nestler suggests that HDACs may be just such a target [32]. The tricyclic antidepressant imipramine increases histone acetylation at specific promoters of the gene encoding BDNF, in part by reducing levels of HDAC 5, a Class 2 HDAC, in the hippocampus. Chronic, but not acute, imipramine reversed the downregulation and overcame hypermethylation of the BDNF locus in response to chronic social defeat stress. Thus, increasing histone acetylation within the hippocampus may reverse the social avoidance observed in mice subjected to chronic stress. This would suggest that HDAC inhibitors might function as antidepressants, or effectively enhance the action of existing antidepressants. Accordingly, the HDAC inhibitor sodium butyrate improves performance in the tail suspension test, a mouse model of antidepressant efficacy, and enhances the efficacy of the selective serotonin reuptake inhibitor fluoxetine [33].
Anxiety disorders
An interesting aspect of anxiety mediated behaviors as well as anxiety disorders is their sensitivity to early life events. In rodents, maternal care stably alters the development of behavioral and endocrine responses to stress in the offspring. Thus, early life experiences alter adult behavior. Such long-lasting changes in behavior appear to be mediated by the epigenetic programming of glucocorticoid receptor gene expression in the hippocampus. In particular, the binding site for the NGFI-A transcription factor in the brain-specific promoter of the glucocorticoid receptor gene promoter is subject to DNA methylation and acetylation. An interesting recent study reveals that treatment of adult offspring with the HDAC inhibitor trichostatin A reverses the effect of maternal care on anxiety related behaviors and differentially affects gene expression of a subset of genes within the hippocampus [34,35]. This is a striking example of the potential therapeutic use of HDAC inhibitors to reverse behavioral alterations that are the result of epigenetic reprogramming during development.
Schizophrenia
Schizophrenia is a serious disorder of cognition with great medical and personal consequence and affects approximately 1.1% of the US population aged 18 and older [36]. Diagnosis is based on simultaneous presentation of a combination of “positive” symptoms (delusions, hallucinations, bizarre thoughts) and negative symptoms (social withdrawal, poor motivation, apathy). Sufferers are unable to function normally in social situations including everyday cognitive tasks. Despite intense research, the molecular etiology of schizophrenia remains enigmatic. Because the symptoms of schizophrenia do not arise until adolescence and persist, in most cases, throughout life, it has been suggested that this disease may be a neurodevelopmental disorder [36].
Two recent findings implicate epigenetic dysfunction in the etiology of schizophrenia. In one study, analysis of postmortem brains from patients diagnosed with schizophrenia or bipolar illness with psychosis showed deficiencies in the extracellular matrix protein reelin [37]. The promoter of reelin contains several sites for DNA methylation and HDAC and DNA methyltransferase inhibitors increase expression of reelin, indicating that epigenetic mechanisms govern the expression of this protein [37]. In subsequent studies, increases in the number of GABAergic mRNA transcripts including glutamic acid decarboxylase 1 (GAD1), a key enzyme for GABA synthesis, were observed in the prefrontal cortex of humans diagnosed with schizophrenia [38,39]. These increases were associated with chromatin remodeling in that an increase in methylation of lysine 4 of histone 3 (H3K4) was observed at GABAergic gene loci over this period of time [38,39]. Although it is as yet too early to know, these observations are consistent with the notion that drugs that target the epigenome are viable therapies for treating this serious mental disorder.
HDAC inhibitors as potential therapeutics for neurodevelopmental disorders
Rubinstein-Taybi Syndrome
Early evidence for a role for epigenetic pathways in brain came from studies showing that the developmental disorders associated with Rubinstein-Taybi Syndrome (RTS) result from heterozygous mutations in the transcriptional coactivator CBP or its homolog p300 [40,41]. RTS is a well-defined inherited, autosomal-dominant disease that occurs once in 125,000 births and accounts for one in 300 patients with mental retardation [42]. Physical traits associated with RTS include altered facial abnormalities, broad digits/fingers and big toes, blunted growth and mental retardation [42].
Several different mouse models of RTS have been created in which CBP or p300 function is genetically altered and these mutant mice exhibit deficits in synaptic plasticity and memory. Mice carrying a single null mutation in the CBP gene, a truncated form of CBP or transgenic mice expressing a dominant negative form of CBP exhibit deficits in long-term memory (LTM), but not in short-term memory (STM) [43–45]. Treatment with the HDAC inhibitors SAHA or TSA ameliorated deficits in synaptic plasticity and cognition in several of the CBP mutant mouse lines, suggesting that even developmental disorders may be amenable to administration in adulthood of therapeutics that target histone acetylation [43,44]. Recent work shows that HDAC inhibitors are only effective if some CREB or CBP function remains [46]. Thus, CBP and CREB appear to recruit HATs to the promoters of specific genes whose expression is altered by HDAC inhibitors to give rise to amelioration of the synaptic and behavioral deficits.
Rett Syndrome
Rett syndrome is also caused by mutations in genes encoding enzymes that mediate chromatin remodeling and affect histone acetylation. Rett syndrome (RS) is a progressive childhood neurodevelopmental disorder that is characterized by mental retardation and stereotypies and arises as a consequence of loss-of-function mutations in the X-linked gene encoding MeCP2 [47–49]. MeCP2 binds to methylated CpG dinucleotides, whereupon it recruits HDACs and transcriptional repressors such as mSin3A. Ultimately, histones associated with 5mCpG become hypoacetylated, promoting tight association between the DNA and the histones and formation of a stable, transcriptionally-repressive chromatin complex [50]. Future studies to explore pharmacological intervention by HDAC inhibitors in the devastating consequences of Rett Syndrome are warranted.
Fragile X syndrome
Fragile X is the most common heritable disorder of mental retardation. It involves a single gene (FMR1) that is transcriptionally silenced as a result of an expanded repeat of CGG trinucleotides (>200) in the 5’-untranslated region (5’-UTR) of the FMR1 gene [51]. The instability of the trinucleotide expansion during embryogenesis can lead to mosaicism in nearly 15% of patients harboring at least 200 CGG repeats. F×S has a prevalence of approximately 1:4000 males and 1:8000 in females. Patients with Fragile X syndrome exhibit a wide-range of neurological deficits including cognitive impairment, seizures, autism, peripheral neuropathy and autonomic dysfunction [51]. The product of the gene, Fragile × Mental Retardation Protein (FMRP) regulates transport of mRNAs into dendrites and local protein synthesis [51]. A pioneering study by Oostra and colleagues showed the ability of the demethylating agent 5-azadeoxycytidine to reactivate FMR1 gene expression in cells derived from patients with Fragile X [52]. Moreover, administration of the HDAC inhibitors 4-phenylbutarate, sodium butyrate or trichostatin in conjunction with 5-azadeoxycytidine was even more effective, indicating synergism between histone hyperacetylation and DNA demethylation [53]. These observations underscore a major role for epigenetic dysregulation in the etiology of Fragile X syndrome.
Future directions and conclusions
Here, we have described a variety of neurodegenerative, psychiatric and neurobehavioral disorde2rs that involve alterations in histone modifications and can be reversed, at least in part, by treatment with HDAC inhibitors. Will HDAC inhibitors treat all ills or will specific classes of HDAC inhibitors be identified to target specific disorders? A common theme that emerges among the diverse neuropsychiatric disorders reviewed in this article is that many of these disorders share a commonality of phenotype as, for example, cognitive deficits. These fundamental clinical features may share common molecular mediators encoded by a subset of interrelated genes. Reduced histone acetylation is a final common endpoint in many of these disorders, but may arise via diverse means including reduced HAT activity, increased HDAC activity or increased DNA methylation (Figure 1). This raises the possibility that new therapeutic strategies used to treat one disorder such as Rubinstein-Taybi Syndrome may also hold promise for treatment of other disorders such as schizophrenia and possibly even neurodegenerative disorders such as Huntington’s disease and ischemia. In this light, “generic” HDAC inhibitors that act throughout the brain might be useful in the amelioration of neurobehavioral defects. Recent evidence reveals that such “generic” HDAC inhibitors have specific effects on gene expression, upregulating a select set of target genes and reducing expression of others [46,54]. These gene-specific effects may be governed by transcription factors, which recognize specific promoter and enhancer motifs and confer target gene specificity and thus sensitivity to HDAC inhibition [46]. From this perspective, then, HDAC inhibitors might selectively reverse expression of only the subset of genes whose expression is altered in a given disorder. An alternative theory posits that more selective HDAC inhibitors might target specific brain regions and act in an isoform-selective and cell-specific manner to reverse disorder-specific epigenetic dysregulation. To date, the HDAC inhibitors used to effectively treat neurodegenerative and psychiatric disorders have been broad-spectrum inhibitors of class I and 2 HDACs (Table 1). In the absence of evidence that the epigenetic dysfunction of any one disorder is mediated by a specific HDAC, it is unclear whether a more selective HDAC inhibitor would be more efficacious. What is clear is that HDAC inhibitors represent a promising new avenue for therapeutic intervention for the cognitive impairments associated with neurodegeneration, chronic stress and neurodevelopmental disorders. Drugs targeted at epigenetic modifications hold the promise of reversing brain disorders once thought to be irreversible.
Figure 1. Scheme showing that neurological and psychiatric disorders involve epigenetic modifications of key neuronal genes and intervention by HDAC inhibitors.
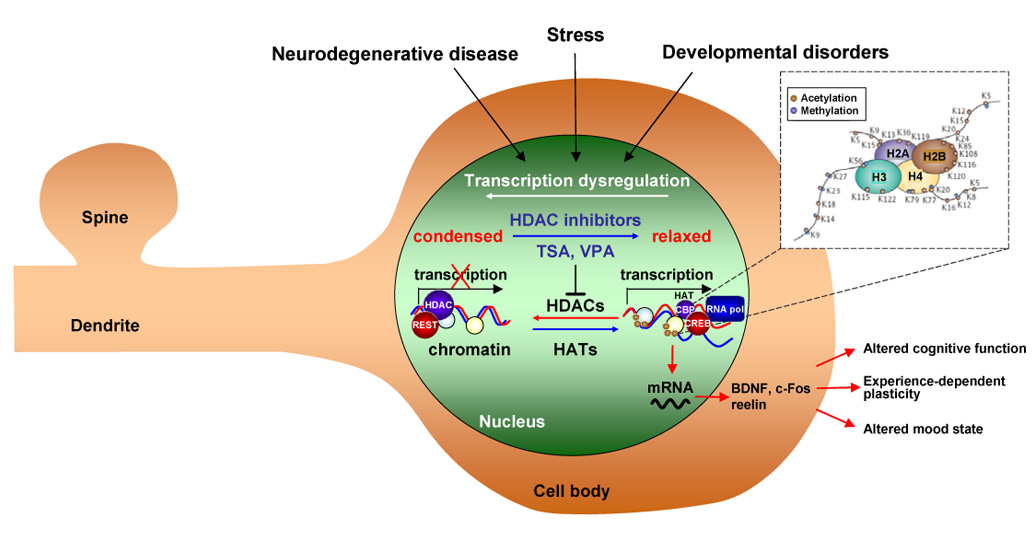
Neurodegenerative diseases (Huntington’s disease, Parkinson’s disease and ischemia), psychiatric disorders (depression, stress and anxiety) and neurodevelopmental disorders (Rubinstein-Taybi syndrome and Fragile X syndrome) can involve aberrant acetylation and methylation of histones and/or DNA methylation. These epigenetic modifications can be influenced by experience and determine the transcriptional state of regulatory genes critical to synaptic plasticity, cognition and mood. Histone deacetylase inhibitors such as valproic acid (VPA) and trichostatin A (TSA) inhibit the activity of HDACs, shifting the balance toward active transcription of neuronal genes and amelioration of plasticity and cognitive deficits. Adapted from [55]. Inset adapted from [8].
Table 1.
Epigenetic basis of neurodegenerative and neuropsychiatric disorders.
| Disorder | Epigenetic Basis | HDAC Inhibitors/ DNA demethylating Agents | HDAC Class | References |
|---|---|---|---|---|
| Huntington’s | histone acetylation ↓ | sodium butyrate, | Class I/II | [12–14] |
| disease | transcriptional | phenylbutyrate | ||
| dysregulation | ||||
| Mouse model of | histone acetylation ↓ | sodium butyrate, | Class I/II | [21] |
| neurodegeneration | trichostatin A | |||
| Stroke | histone acetylation ↓ | SAHA, 5-aza-2’- | Class I/II | [28,29] |
| DNA methylation ↑ | deocycytidine | |||
| Depression | histone acetylation ↓ | sodium butyrate | Class I/II | [32,33] |
| Schizophrenia | H3K4 methylation ↑ | 5-aza-2’-deoxycytidine? | [37–39] | |
| DNA methylation ↑ | ||||
| Rubinstein-Taybi | histone acetylation ↓ | SAHA, trichostatin A | Class I/II | [43–46] |
| Syndrome | ||||
| Fragile X | DNA methylation ↑ | sodium butyrate or TSA | Class I/II | [52,53] |
| + 5-aza-2’-deoxycytidine |
Acknowledgments
This work was supported by NIH grant NS46742 to RSZ, NIH grant MH60244 to TA and a generous grant from the F. M. Kirby Program in Neural Repair and Neuroprotection to RSZ. The authors declare no conflicting financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 3.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 4.Wood MA, Hawk JD, Abel T. Combinatorial chromatin modifications and memory storage: a code for memory? Learn Mem. 2006;13:241–244. doi: 10.1101/lm.278206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- 6.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 7.Carey N, La Thangue NB. Histone deacetylase inhibitors: gathering pace. Curr Opin Pharmacol. 2006;6:369–375. doi: 10.1016/j.coph.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 8.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 9.Bates GP. Huntington's disease. Exploiting expression. Nature. 2001;413:691, 693–691, 694. doi: 10.1038/35099656. [DOI] [PubMed] [Google Scholar]
- 10.Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- 11.Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- ••12.Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••13.Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ, Beal MF. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington's disease. J Biol Chem. 2005;280:556–563. doi: 10.1074/jbc.M410210200. [DOI] [PubMed] [Google Scholar]
- ••14. Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci U S A. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. ••These three papers convincingly document the ability of HDAC inhibitors to ameliorate neurodegeneration and/or behavioral deficits in a mouse model of Huntington’s disease.
- 15.Gasser T. Genetics of Parkinson's disease. J Neurol. 2001;248:833–840. doi: 10.1007/s004150170066. [DOI] [PubMed] [Google Scholar]
- 16.Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15:3012–3023. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- 17.Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao X, Sudhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 19.Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, Teplow DB, Haass C. Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2001;2:835–841. doi: 10.1093/embo-reports/kve180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selkoe DJ. Presenilin, Notch, and the genesis and treatment of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:11039–11041. doi: 10.1073/pnas.211352598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- 22.Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 23.Ballas N, Mandel G. The many faces of REST oversee epigenetic programming of neuronal genes. Curr Opin Neurobiol. 2005;15:500–506. doi: 10.1016/j.conb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 24.Ooi L, Wood IC. Chromatin crosstalk in development and disease: lessons from REST. Nat Rev Genet. 2007;8:544–554. doi: 10.1038/nrg2100. [DOI] [PubMed] [Google Scholar]
- 25.Roopra A, Huang Y, Dingledine R. Neurological Disease: Listening to Gene Silencers. Mol Interv. 2001;1:219–228. [PubMed] [Google Scholar]
- 26.Calderone A, Jover T, Noh K-M, Tanaka H, Yokota H, Lin Y, Grooms S, Regis R, Bennett MV, Zukin RS. Ischemic insults de-repress the gene silencer rest in neurons destined to die. J Neurosci. 2003;23:2112–2121. doi: 10.1523/JNEUROSCI.23-06-02112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••27. Formisano L, Noh KM, Miyawaki T, Mashiko T, Bennett MV, Zukin RS. Ischemic insults promote epigenetic reprogramming of mu opioid receptor expression in hippocampal neurons. Proc Natl Acad Sci USA. 2007;104:4170–4175. doi: 10.1073/pnas.0611704104. ••This is the first study to document epigenetic remodeling of gene expression in an experimental model of global ischemia in rat. The study demonstrates that global ischemia promotes deacetylation of H3K9, but not H3K4 over the promoter of the mu opiate receptor and further demonstrates that suppression of mu receptor expression is neuroprotective.
- ••28. Faraco G, Pancani T, Formentini L, Mascagni P, Fossati G, Leoni F, Moroni F, Chiarugi A. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol Pharmacol. 2006;70:1876–1884. doi: 10.1124/mol.106.027912. ••This paper convincingly documents the ability of HDAC inhibitors to ameliorate neurodegeneration and ischemic injury in a mouse model of focal ischemia.
- ••29. Endres M, Meisel A, Biniszkiewicz D, Namura S, Prass K, Ruscher K, Lipski A, Jaenisch R, Moskowitz MA, Dirnagl U. DNA methyltransferase contributes to delayed ischemic brain injury. J Neurosci. 2000;20:3175–3181. doi: 10.1523/JNEUROSCI.20-09-03175.2000. ••This paper convincingly documents the ability of demethylating agents to ameliorate neurodegeneration and ischemic injury in a mouse model of focal ischemia.
- 30.Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- 31.Hyman SE. Even chromatin gets the blues. Nat Neurosci. 2006;9:465–466. doi: 10.1038/nn0406-465. [DOI] [PubMed] [Google Scholar]
- ••32.Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- ••33. Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55–64. doi: 10.1016/j.biopsych.2006.06.036. ••These papers describe the role of chromatin modifications in mouse models of depression and document the potential antidepressant-like properties of HDAC inhibitors.
- 34.Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 35.Weaver IC, Meaney MJ, Szyf M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci U S A. 2006;103:3480–3485. doi: 10.1073/pnas.0507526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sawa A, Snyder SH. Schizophrenia: diverse approaches to a complex disease. Science. 2002;296:692–695. doi: 10.1126/science.1070532. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y, Sharma RP, Costa RH, Costa E, Grayson DR. On the epigenetic regulation of the human reelin promoter. Nucleic Acids Res. 2002;30:2930–2939. doi: 10.1093/nar/gkf401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang HS, Matevossian A, Whittle C, Kim SY, Schumacher A, Baker SP, Akbarian S. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci. 2007;27:11254–11262. doi: 10.1523/JNEUROSCI.3272-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang HS, Akbarian S. GAD1 mRNA expression and DNA methylation in prefrontal cortex of subjects with schizophrenia. PLoS ONE. 2007;2:e809. doi: 10.1371/journal.pone.0000809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blough RI, Petrij F, Dauwerse JG, Milatovich-Cherry A, Weiss L, Saal HM, Rubinstein JH. Variation in microdeletions of the cyclic AMP-responsive element-binding protein gene at chromosome band 16p13.3 in the Rubinstein-Taybi syndrome. Am J Med Genet. 2000;90:29–34. doi: 10.1002/(sici)1096-8628(20000103)90:1<29::aid-ajmg6>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 41.Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 42.Weeber EJ, Levenson JM, Sweatt JD. Molecular genetics of human cognition. Mol Interv. 2002;2:376–391. 339. doi: 10.1124/mi.2.6.376. [DOI] [PubMed] [Google Scholar]
- ••43.Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP +/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- ••44.Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••45. Bourtchouladze R, Lidge R, Catapano R, Stanley J, Gossweiler S, Romashko D, Scott R, Tully T. A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc Natl Acad Sci U S A. 2003;100:10518–10522. doi: 10.1073/pnas.1834280100. ••These three papers convincingly demonstrate the ability of HDAC inhibitors to afford amelioration of synaptic plasticity and cognition in mouse models of Rubinstein-Taybi Syndrome, an autism spectrum disorder.
- ••46. Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. ••This paper identifies the mechanisms by which HDAC inhibitors enhance synaptic plasticity and memory and begins to define the critical genes whose expression is regulated by histone acetylation during memory storage.
- 47.Amir RE, Van dV, I, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 48.Hagberg B. The Rett syndrome: an introductory overview 1990. Brain Dev. 1992;14 Suppl:S5–S8. [PubMed] [Google Scholar]
- 49.Olsson B, Rett A. A review of the Rett syndrome with a theory of autism. Brain Dev. 1990;12:11–15. doi: 10.1016/s0387-7604(12)80166-5. [DOI] [PubMed] [Google Scholar]
- 50.Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- 51.O'Donnell WT, Warren ST. A decade of molecular studies of fragile X syndrome. Annu Rev Neurosci. 2002;25:315–338. doi: 10.1146/annurev.neuro.25.112701.142909. [DOI] [PubMed] [Google Scholar]
- ••52.Chiurazzi P, Pomponi MG, Willemsen R, Oostra BA, Neri G. In vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum Mol Genet. 1998;7:109–113. doi: 10.1093/hmg/7.1.109. [DOI] [PubMed] [Google Scholar]
- ••53. Chiurazzi P, Pomponi MG, Pietrobono R, Bakker CE, Neri G, Oostra BA. Synergistic effect of histone hyperacetylation and DNA demethylation in the reactivation of the FMR1 gene. Hum Mol Genet. 1999;8:2317–2323. doi: 10.1093/hmg/8.12.2317. ••These two papers convincingly demonstrate the ability of the demethylating agent 5-azad-d alone or in conjunction with HDAC inhibitors to promote reactivation of the FMR1 gene in cells derived from patients with Fragile X Syndrome, the most prevalent form of inherited mental retardation.
- 54.Fass DM, Butler JE, Goodman RH. Deacetylase activity is required for cAMP activation of a subset of CREB target genes. J Biol Chem. 2003;278:43014–43019. doi: 10.1074/jbc.M305905200. [DOI] [PubMed] [Google Scholar]
- 55.Duman RS, Newton SS. Epigenetic marking and neuronal plasticity. Biol Psychiatry. 2007;62:1–3. doi: 10.1016/j.biopsych.2007.04.037. [DOI] [PubMed] [Google Scholar]