

Abstract
The protooncogene c-abl encodes a nonreceptor tyrosine kinase whose cellular function is unknown. To study the possible involvement of c-Abl in proliferation, differentiation, and cell cycle regulation of early B cells, long-term lymphoid bone marrow cultures were established from c-abl-deficient mice and their wild-type littermates. Interleukin 7-dependent progenitor B-cell clones and lines expressing B220 and CD43 could be generated from both mutant and wild-type mice. The mutant and wild-type lines displayed no difference in their proliferative capacity as measured by thymidine incorporation in response to various concentrations of interleukin 7. Similarly, c-abl deficiency did not interfere with the ability of mutant clones to differentiate into surface IgM-positive cells in vitro. Analysis of cultures after growth factor deprivation, however, revealed a strikingly accelerated rate of cell death in c-abl mutant cells, due to apoptosis as confirmed by terminal deoxynucleotidyltransferase-mediated UTP nick end labeling analysis. Furthermore, a greater susceptibility to apoptotic cell death in c-abl mutant cells was also observed after glucocorticoid treatment. These results suggest that mutant c-Abl renders the B-cell progenitors more sensitive to apoptosis, and may account for some of the phenotypes observed in c-abl-deficient animals.
Keywords: tyrosine kinase, Whitlock–Witte cultures, interleukin 7, apoptosis
The Abelson murine leukemia virus (1) contains a single gene, v-abl, that is responsible for the oncogenic transformation of early B-cell progenitors (2). The viral gene, encoding a potent tyrosine kinase, is an activated version of a cellular protooncogene termed c-abl. Human oncogenic variants of c-Abl (p185 BCR-ABL and p210 BCR-ABL) can also be generated by chromosomal translocation, and are critically involved in the development of chronic myelogenous leukemia and certain acute lymphoid leukemias (2, 3). In oncogenic variants of Abl, the tyrosine kinase is constitutively activated and the predominantly nuclear localization of c-Abl is altered to a cytoplasmic localization (2). v-Abl as well as BCR-ABL can abrogate the growth factor requirement in factor-dependent cell lines and transform fibroblasts (2, 4). v-Abl promotes cell cycle progression in early B cells (5), and inhibits light chain immunoglobulin rearrangement and B lymphoid differentiation (6, 7). Both v-Abl and BCR-ABL have been shown to be able to prevent apoptosis (8–12), providing an alternative or additional mode for their transforming capacity.
Whereas the transforming mechanism of v-Abl and BCR-ABL have been extensively characterized, the biological function of c-Abl is less well-understood. The various roles of c-Abl in cell physiology may or may not be simply related to the effects of v-Abl and BCR-ABL activity. Mice deficient for the C terminus of c-Abl or for the complete c-Abl protein have been generated by targeted gene disruption, and both show a variety of defects including high postnatal mortality, runting, and abnormalities in lymphoid cells such as splenic and thymic atrophy and lymphopenia (13, 14). Characterization of the lymphoid compartment of the abl-deficient mice revealed reduced numbers of thymocytes and B-cell progenitors in some of the mice and a perturbed response to interleukin 7 (IL-7) and bacterial lipopolysaccharide (13–16). All the phenotypes of the mutant animals were highly variable, with some animals being nearly normal and others profoundly defective. Transfer of bone marrow from mutant mice to irradiated syngeneic wild-type mice reproduced the B-cell population defect in the recipient animals (13, 15), suggesting that the defect is cell-autonomous.
Several recent studies have implicated c-Abl in cell cycle regulation, but so far no detailed picture of its mechanism of action has emerged. Overexpression of c-Abl in NIH 3T3 cells induces G1 arrest, indicating a negative regulatory role for c-Abl in cell growth (17). This effect was shown to be dependent on p53 but not the retinoblastoma protein (RB) (18). On the other hand, a positive role for c-Abl in cell growth was suggested by the observation that overexpression of c-Abl can overcome RB-induced growth arrest in osteosarcoma cells (19). Moreover, down-regulation of c-Abl with antisense oligodeoxynucleotides or antisense RNA led to inhibition of cell growth in CD34+ hematopoietic cells (20) or NIH 3T3 cells (21). c-Abl may thus play either growth-promoting or growth-arresting roles in different settings.
Here we report on the generation and functional characterization of progenitor B-cell lines of abl-deficient mice. These cell lines provide an experimental system in which to study the possible involvement of c-Abl in proliferation, cycle regulation, and differentiation. Examination of these cells has revealed a significant sensitivity to premature apoptotic cell death, potentially accounting for the lymphopenia manifested by the mutant animals.
MATERIALS AND METHODS
Growth Factors, Antibodies, and Animals.
Recombinant mouse IL-7 was purchased from R & D Systems. Supernatant culture medium from the TSA-IL7 cell line (22) was used at a concentration of 5% as a source of recombinant mouse IL-7 to maintain progenitor B-cell cultures. Monoclonal rat anti-mouse antibodies against heavy-chain [331 (23)], CD43 [S7 (24)], and Mac-1 [M1/70 (25)] were kindly provided by Alan Stall (Columbia University). Avidin-Texas red and antibodies against B220 [RA3-6B2 (26)] were purchased from PharMingen. Ablm1 mice (129/Sv/Ev background) were bred in a specific pathogen-free animal facility and genotyped as previously described (13).
Generation of Progenitor B-Cell Clones and Lines.
Bone marrow from 4- to 6-week-old ablm1 mice and control littermates was isolated and cultured at 1 × 106 cells per ml in RPMI 1640 medium supplemented with 5% fetal calf serum (FCS, HyClone) and 50 μM 2-mercaptoethanol according to the method of Whitlock and Witte (27). After a culture period of 4–5 weeks, nonadherent cells were collected. Part of the nonadherent cell suspension was cloned by limiting dilution at 0.5 cells per well on the stromal cell line ST-2 (28) in the presence of IL-7 in a 96-well microtiter plate. The remaining cell suspension was cultured with IL-7 in the absence of a stromal layer (10% FCS instead of 5%). ST-2 cells did not produce IL-7 as measured by a bioassay using IL-7-dependent IXN2B cells (29). The following nomenclature was used: AP stands for abl-positive, AN stands for abl-negative cultures. Clones and lines generated from littermates have the same Arabic number. Bone marrow of five homozygous and four control mice were used to generate the lines and clones described here.
Proliferation Assay.
Cells were cultured at a density of 5 × 103 per 200 μl in a 96-well round-bottom microtiter plate with varying concentrations of recombinant mIL-7 in culture medium for 72 h. During the last 6 h of culture, cells were pulse-labeled with 0.5 μCi of [3H]thymidine (5 Ci/mmol; 1 Ci = 37 GBq; Amersham), and [3H]thymidine incorporation was quantified by scintillation counting as described (29).
Assays for Cell Death and Apoptosis.
Cells were washed three times in PBS to remove IL-7 and then resuspended in RPMI 1640 medium supplemented with 10% FCS and 50 μM 2-mercaptoethanol at 1 × 106 cell per ml. At different times after IL-7 withdrawal, samples were taken and the percentage of viable cells was determined by trypan blue exclusion. To determine the percentage of apoptotic cells, 1 × 106 cells were labeled after 15 h by terminal deoxynucleotidyltransferase-mediated UTP nick end labeling (TUNEL) using the in situ cell death detection kit (Boehringer Mannheim) according to the manufacturer’s instructions and analyzed with a fluorescence-activated cell sorter.
Cell Cycle Analysis.
Cell cycle analysis was done as described using a modification of the 5-bromo-2′-deoxyuridine (BrdU) labeling and detection kit I from Boehringer Mannheim (30). Cells were labeled with BrdU (10 μmol/liter) for 30 min at a cell concentration of 2 × 106 cells per ml, washed once in PBS, and fixed in 70% ethanol (in 50 mM glycine buffer, pH 2.0) for at least 24 h. After fixation, cells were washed twice in PBS and then incubated in 200 μl of anti-BrdU solution at 37°C for 1 h. Cells were washed twice in PBS and resuspended in 200 μl of anti-mouse Ig-fluorescein solution. After a 30-min incubation at 37°C in the dark, the cells were washed twice in PBS and resuspended in 500 μl of PBS containing 50 μg of propidium iodide per ml. Cells were analyzed with a fluorescence-activated cell sorter.
RESULTS
Generation of Progenitor B-Cell Lines and Clones.
Long-term bone marrow cultures from c-abl-deficient mice and their wild-type littermates were established. After 4–5 weeks, nonadherent cells were collected and cultured according to one of two protocols. In one set of cultures, clones were established by limiting dilution on the stroma cell line ST-2 in the presence of IL-7; this protocol provides both cell contact-dependent stimulation and soluble factor stimulation. In another set, cell lines were established by culture in the presence of IL-7 only, without cloning. Stroma- and IL-7-dependent cell clones and IL-7-dependent lines could be generated from wild-type as well as abl-deficient mice under both conditions.
To characterize the developmental stages of the cultures, surface expression of CD43, B220, and IgM by the cells was measured by flow cytometry (Tables 1 and 2) as described previously (15). In both mutant and wild-type backgrounds, a variety of different phenotypes were observed among the various cultures. All clones and lines expressed CD43. Many of the clones and lines expressed B220, but its expression could not be detected on about half of the clones, and on the single line AP-3. Surface IgM was expressed on only two of the lines, AN-2 and AP-3. None of these markers correlated in any distinctive way with the abl-deficient genotype of the donor animals.
Table 1.
Surface markers and genotype of IL-7-dependent progenitor B-cell lines that grow independent of stroma
| Cell line | Wild-type abl | Surface expression
|
||
|---|---|---|---|---|
| CD43 | B220 | IgM | ||
| AN-1 | − | + | + | − |
| AP-1 | + | + | + | − |
| AN-2 | − | + | + | + |
| AP-2 | + | + | + | − |
| AN-3 | − | + | + | − |
| AP-3* | + | + | − | + |
Heterozygous for the mutant abl allele.
Table 2.
Surface markers and genotype of IL-7- and stroma-dependent progenitor B-cell clones
| Clone | Wild-type abl | Surface expression
|
|||
|---|---|---|---|---|---|
| CD43 | B220 | IgM | IgM−IL-7* | ||
| AN-4a | − | + | + | − | − |
| AN-4b | − | + | − | − | 4.7% |
| AN-4c | − | + | + | − | − |
| AP-4a | + | + | − | − | − |
| AP-4b | + | + | + | − | 6% |
| AN-5 | − | + | + | − | 3.1% |
| AP-5 | + | + | − | − | 6.4% |
| AN-6 | − | + | − | − | 3.6% |
| AN-7 | − | + | − | − | − |
| AP-8† | + | + | + | − | − |
IgM expression 3 days after IL-7 withdrawal.
Heterozygous for the mutant abl allele.
The expression of CD43 and B220 in the absence of surface IgM chain, as seen on most of the cultures, and the IL-7- and stroma-dependence, are indicative of a pro-B-cell stage (31). The IL-7-dependent cell lines that grew without stromal cells seem to represent a transitional stage in which the stromal cell-dependence is lost but the IL-7-dependence is retained. A similar phenotype has been described by others (32–35) and probably corresponds to a late pro-B- or early pre-B-cell stage. The surface IgM expression on AN-2 and AP-3 is unusual because expression of surface Ig is usually correlated with loss of IL-7-dependence, but some similar cell lines have been reported (32, 34). The lack of B220 expression on some of the clones and on line AP-3 might be due to loss of the B220 epitope from an initially B220-positive precursor, as has been described for other IL-7-dependent B-cell progenitors after prolonged culture (36). Most of the cells that lack B220 are clearly committed to the B-cell lineage, as is made evident by the expression of IgM on AP-3 or the inducible expression of IgM on the clones (see below).
The properties of the cultures recovered in these experiments suggest that abl-deficiency did not hamper the generation of B-cell progenitor clones and lines at early stages of B-cell development. No significant differences in the expressed surface markers were observed between abl-deficient and wild-type clones and lines.
In Vitro Differentiation of Progenitor B Cells to Surface IgM-Positive B Cells.
Withdrawal of IL-7 from IL-7- and stroma-dependent progenitor B cells first induces their differentiation into VHDHJH/VLJL-rearranged, surface immunoglobulin-positive B cells, and subsequently induces apoptotic cell death (32). To determine if c-Abl is required for the induction of surface IgM, the mutant and wild-type clones were analyzed for their potential to differentiate in vitro. Initially, IgM-negative progenitor B-cell clones were cultured for 3 days on a stromal cell line in the absence of IL-7 and then analyzed for surface expression of heavy chain by flow cytometry. A small but significant number of wild-type and abl-deficient progenitor B-cell clones reacted to IL-7 withdrawal by surface expression of heavy chain (Fig. 1 and Table 2). There was no significant difference between mutant and wild-type cultures in the number of cells becoming surface IgM-positive. The low percentage of cells showing expression of IgM is in agreement with observations made by others (32), and may reflect the naturally low frequency of productive rearrangements. The data suggest that functional c-Abl is not required for pro-B cells to undergo differentiation to a surface IgM-positive stage in vitro.
Figure 1.
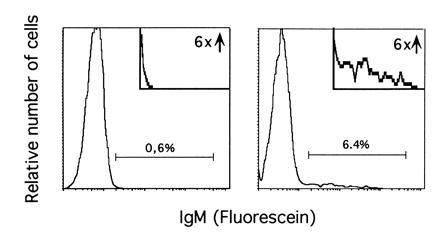
Example of the analysis of surface IgM expression after IL-7 withdrawal. Clone AP-5 was cultured on the stromal layer for 3 days in the presence (Left) or in the absence (Right) of IL-7. Fluorescence-activated cell sorter analysis of IgM expression was performed as described. (Insets) An enlargement of the areas containing IgM-positive cells.
Proliferative Response of abl-Deficient Progenitor B-Cell Lines to IL-7 Is Unaltered.
Previous studies have revealed that the ability to form colonies in the presence of IL-7 in a semisolid matrix (CFU-IL-7) is often perturbed in c-abl-deficient bone marrow, raising the possibility that c-Abl might play a role in IL-7 signaling (15). We compared the proliferative response of mutant and wild-type stroma-independent progenitor B-cell lines to different concentrations of IL-7 by measuring thymidine incorporation over the course of 3 days (Fig. 2). The dose-response curve of mutant and wild-type cell lines was nearly indistinguishable over a wide range of IL-7 concentrations (0.03–50 ng/ml). This indicates that the abl-deficient lines have no intrinsic defect in their proliferative response to IL-7. Thus, the cell lines do not show an IL-7 nonresponsive behavior that would account for the deficiency in vivo (15).
Figure 2.

Comparison of the proliferative response of abl-deficient and control lines to IL-7. [3H]Thymidine incorporation as an indicator of cellular proliferation is plotted against rising IL-7 concentrations. Cells were cultured for 72 h with recombinant IL-7 and pulse labeled with [3H]thymidine during the last 6 h of culture (see Materials and Methods). The IL-7 dose-response curves of the abl-deficient clones AN-1, AN-2, and AN-3 and the control clones AP-1, AP-2, and AP-3 are indistinguishable.
Cell Cycle Analysis.
Several recent studies have implicated c-Abl in cell cycle regulation, but they have reached contrasting results concerning the question of whether c-Abl is a positive or negative regulator of cell growth (17, 19–21). To test for alterations in cell cycle progression, we analyzed the distribution of wild-type and mutant stroma-independent cells in the different cell cycle phases under normal logarithmic growth in the presence of IL-7. Cells were labeled with BrdU (as a marker of DNA synthesis) and propidium iodide (as a marker of DNA content) and both parameters were analyzed simultaneously with a fluorescence-activated cell sorter. Plotting of the DNA content against DNA synthesis allows differentiation of cells in the G0/G1, S, and G2/M phases. Fig. 3 Left shows the cell cycle profiles of a mutant (AN-3, Fig. 3a) and a wild-type (AP-2, Fig. 3d) line, representative of the profiles obtained with the four other lines. Comparison of wild-type and mutant cells revealed a similar distribution among the G0/G1, S, and G2/M phases, indicating that the mutant Abl did not interfere with normal cell cycle progression. The low percentage of cells in the G2/M phase is consistent with results obtained by others for progenitor B cells (36).
Figure 3.
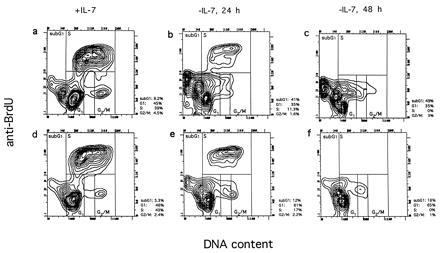
Cell cycle profiles of an abl-deficient (a–c) and a control line (d–f) in the presence of IL-7 and after IL-7 withdrawal for 24 (b and e) and 48 h (c and f). Cells were labeled with BrdU during the last 30 min of culture and subsequently stained with anti-BrdU antibodies and propidium iodide and analyzed by fluorescence-activated cell sorter (see Materials and Methods). The percentage of cells in G0/G1, S, G2/M, and sub-G1 are shown to the right of each plot. The cell population that occurs after IL-7 withdrawal to the right of the G1 population and stretches toward the G2/M population may represent cells that underwent apoptosis in S or G2/M. Because they extend into the G2/M and G1 gate, the percentages for G2/M and G1 may be overestimated.
IL-7-dependent progenitor B cells have been shown to undergo G1 arrest following IL-7 withdrawal (36). To study the ability of the mutant and wild-type cells to undergo G1 arrest, we analyzed the cell cycle distribution at different times (24 and 48 h) after complete IL-7 withdrawal. Growth factor withdrawal from wild-type cultures resulted in the accumulation of the cells in the G0/G1 phase and slight accumulation of cells with a lower DNA content than the G0/G1 population (sub-G1 region) (Fig. 3 e and f). This population probably represents apoptotic cells that have a reduced DNA content due to the leakage of DNA fragments (37). The vast majority of the IL-7-deprived wild-type cells accumulated predominantly in G1, and showed only a moderate increase of cells in sub-G1 over time. This accumulation of cells in the G0/G1 phase is indicative of a G1 arrest.
Examination of the mutant cell lines (Fig. 3 b and c) revealed a very different behavior upon IL-7 withdrawal. Here much less accumulation in G0/G1 was observed: after 48 h, only 35% of the abl-deficient cells were detected in G0/G1, compared with 65% of the wild-type cells. Instead, most of the abl-deficient cells were found in the sub-G1 region after 24 (Fig. 3b) and 48 h (Fig. 3c), suggesting a strongly enhanced apoptotic cell death. After 48 h, 49% of the abl-deficient cells were in the sub-G1 region, compared with 18% of the wild-type cells. Analysis of the other abl-deficient (AN-1 and AN-2) and control lines (AP-1 and AP-3) confirmed the observations made in AN-3 and AP-2, respectively (data not shown). These data suggest that there is a striking difference in the response to cytokine withdrawal in cells deficient in c-Abl.
Mutant c-abl Renders Progenitor B-Cell Lines and Clones More Susceptible to Apoptotic Stimuli.
To study apoptotic cell death in the abl-deficient cell lines directly, we determined the rate of cell death after withdrawal of IL-7. Fig. 4 shows the percentage of viable cells as determined by trypan blue exclusion at different times after IL-7 withdrawal. Abl-deficient cell lines consistently showed a higher rate of cell death than wild-type lines.
Figure 4.
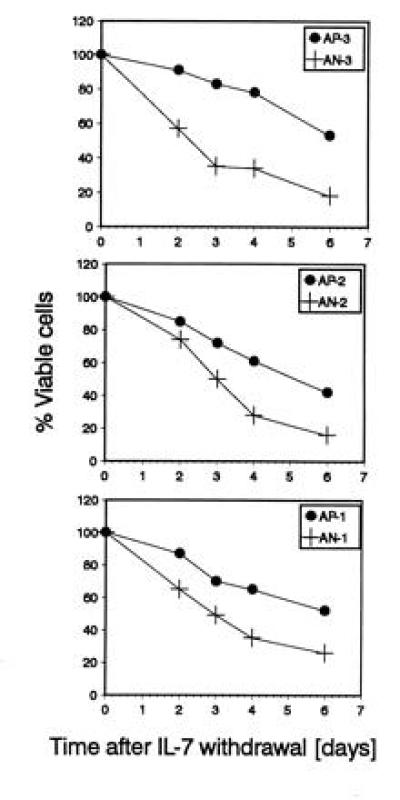
Viability of abl-deficient and control lines after IL-7 deprivation. The percentage of viable cells was determined by trypan blue exclusion at different time points after IL-7 withdrawal. Each blot compares the viability of an abl-deficient line to a control line generated from littermates. Abl-deficient lines (+) show a lower viability than their control lines (•). For each data point, duplicate cultures were analyzed. The SD was ≤5%. One of two experiments with similar results is shown.
Cell death can occur by either apoptosis or necrosis; apoptotic cells break down high molecular weight DNA into shorter fragments. We used the TUNEL method (38) to determine whether the observed cell death was due to apoptosis and to quantify the amount of apoptotic cells after IL-7 withdrawal. Terminal deoxynucleotidyltransferase labels the ends of breakdown fragments with fluorescein-conjugated dUTP and allows the detection and quantitation of apoptotic cells by fluorescence-activated cell sorter analysis. Cell lines were IL-7-deprived for 15 h and then analyzed by the TUNEL method. Fig. 5 shows that the percentage of apoptotic cells as detected by fluorescein staining is about twice as high in abl-deficient lines than in wild-type lines. The same analysis was also performed with the IL-7- and stroma-dependent progenitor B-cell clones. We observed 30% more apoptotic cells in abl-deficient clones than in wild-type clones (data not shown). This difference was less pronounced than for the stroma-independent lines. Nevertheless, it confirmed the increased apoptotic sensitivity of abl-deficient cells.
Figure 5.

Increased cell death in abl-deficient cell lines after IL-7 deprivation is due to apoptosis. Cells were cultured in the absence of IL-7 for 15 h and then assayed for apoptosis by TUNEL analysis (see Materials and Methods). A higher percentage of apoptotic cells was detected in abl-deficient lines (Left) compared with control lines (Right).
Finally, we analyzed whether the increased apoptotic cell death in abl-deficient cells could be reproduced with another apoptotic stimulus. Cells were treated with 1 μM dexamethasone for 8 h and the percentage of living cells was determined by trypan blue exclusion. As shown in Fig. 6, dexamethasone-treated abl-deficient cells showed a higher proportion (2- to 4-fold) of cell death than identically treated wild-type cells. We confirmed by TUNEL analysis that the cell death was due to apoptosis (data not shown). The results show that the lack of a functional c-abl allele renders cells more susceptible to two different apoptotic stimuli, and suggest that the wild-type c-Abl acts to prevent apoptosis.
Figure 6.
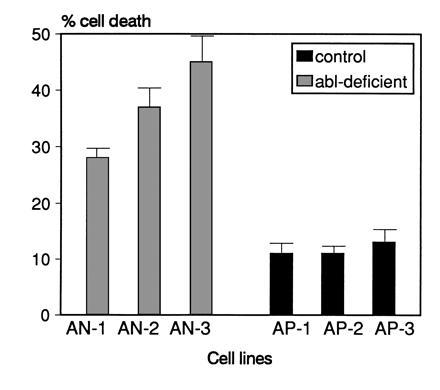
Rate of cell death after dexamethasone treatment is increased in abl-deficient lines. Cells were treated with 1 μM dexamethasone for 8 h and the percentage of dead cells was determined by trypan blue exclusion. For each data point, duplicate cultures were analyzed. Error bars indicate SD values. One of two experiments with similar results is shown.
DISCUSSION
By several criteria, progenitor B-cell lines and clones generated from abl-deficient mice were very similar to those obtained from wild-type mice. Analysis of surface markers displayed by various clones and lines revealed no significant difference in the spectrum of phenotypes recovered from abl-deficient and wild-type animals (Tables 1 and 2). Although the abl-deficient animals display deficiencies in their progenitor B-cell populations in some assays (13, 15), the mutant bone marrow must contain sufficient numbers of precursors to allow in vitro expansion to form lines and clones. This result is consistent with the previous observation that long-term bone marrow cultures are normal in abl-deficient mice (15). Furthermore, the proliferative response of the matched cell lines to IL-7 and their cell cycle progression in the presence of IL-7 was similar (Figs. 2 and 3), indicating that the perturbed pre-B-cell compartment in abl-deficient mice is not caused by a general interference with the IL-7 signaling pathway. Our experiments also failed to demonstrate an influence of abl-deficiency on progenitor B-cell differentiation in vitro, since abl-deficient clones were capable of normal differentiation to the stage of surface IgM expression after IL-7 withdrawal (Tables 1 and 2). Thus, these aspects of the cultures do not shed light on what could account for the lymphopenia and functional defects seen in the c-abl mutant mice (15).
Whereas proliferation, cell cycle progression, and differentiation into IgM+ cells seemed to be unimpaired by the mutant abl allele, we observed a significantly increased sensitivity of abl-deficient cells to apoptotic stimuli. Growth factor withdrawal as well as dexamethasone treatment induced a higher rate of cell death in abl-deficient cells compared with wild-type cells (Figs. 4 and 6). The observed increased susceptibility to apoptosis may explain some of the observations made during the characterization of abl-deficient mice. In the bone marrow of abl-deficient mice, a reduction of pre- and pro-B cells was noted (13). In a short-term in vitro B cell-lymphopoiesis culture system, a reduction of pro-B cells in the cultures of abl mutant mice was observed after 6 days, whereas the numbers of more mature B cells were not reduced (15). Our data suggest that the reduced number of early B cells may have been due to increased apoptotic cell death. IL-7-dependent B-cell progenitors were recently shown to be very sensitive to apoptosis (39), and we would suggest that the c-abl-deficiency can enhance this sensitivity.
One of the most puzzling aspects of the c-abl-deficient phenotype is the profound variability from animal to animal in the severity of the defects. Although the numbers of pro- and pre-B cells are profoundly reduced in some c-abl mutant animals, others show nearly normal levels (13, 15). The sensitivity of the mutant lines to apoptotic death might explain the high variability of the phenotype seen in different mice. Environmental stress is a significant variable in the maintenance of laboratory animals, and glucocorticoid levels are known to vary in response to such stress. Glucocorticoids have been shown to exert profound effects on the numbers of B lineage precursors even in wild-type animals (39–41). These effects might be enhanced in abl-deficient mice due to the increased sensitivity of their B-cell progenitors to glucocorticoid-induced apoptosis.
The mechanism of increased susceptibility to apoptosis in abl-deficient cells remains to be analyzed. Several molecules have been shown to be involved in the regulation of apoptosis following growth factor withdrawal (42). In some cell types, overexpression of Bcl-2 has been shown to prevent cell death induced by growth factor withdrawal (42, 43), inactivation of v-abl (6, 7), or glucocorticoid treatment (39). Moreover, Bcl-2 seems to mediate the inhibition of apoptosis induced by expression of BCR-ABL in IL-3-dependent cell lines (11). This raises the possibility that Bcl-2 might also be a downstream mediator of c-Abl’s normal involvement in reducing apoptosis.
It is possible that the increased apoptosis observed in abl-deficient cells is linked to deregulation of the cell cycle. The relationship between cell cycle regulation and apoptosis is complex and remains poorly understood (42, 44). Failure to arrest at check points, and entry into cell cycle under inappropriate conditions can lead to apoptosis (45–47). On the other hand, apoptosis appears to be closely linked to the interruption of the cell cycle stimulation after growth factor withdrawal (42, 43). Our data do not allow us to conclude that there is an alteration of the cell cycle in abl-deficient cells or whether the observed changes in the cell cycle distribution of the cells were secondary to the apoptosis: control cells but not abl-deficient cells accumulated in G0/G1, but we cannot determine whether abl-deficient cells failed to accumulate in G0/G1 because they became apoptotic more rapidly or they became apoptotic because they failed to be arrested in G1. Experiments are under way to analyze the cell cycle check points in more detail in the presence of genes that may prevent the apoptotic response (e.g., Bcl-2).
It is tempting to speculate that the increased susceptibility to apoptotic stimuli we observe in abl-deficient progenitor B cells reflects part of its elusive physiological function. Only a very small fraction of the B cells generated in the bone marrow eventually survive. These early B cells have been shown to be exquisitely sensitive to apoptosis, which may be explained by strongly decreased expression of Bcl-2 (48). Abl may play a negative regulatory role in the fine-tuning of the apoptotic susceptibility that is important in the decision to make most cells die and some survive. The cell lines described here provide an experimental means to define the role of c-Abl in apoptosis on a molecular level.
Finally, we note that since c-Abl is ubiquitously expressed (2), the antiapoptotic function of Abl may not be limited to early B cells. Defects in apoptosis may account for other elements in the pathology of abl-deficient mice that are poorly understood: premature death, thymic atrophy, runting, and abnormalities in the gastrointestinal tract (13, 14).
Acknowledgments
We are grateful to Sharon Boast and Kenia de los Santos for excellent technical assistance. We also thank Dr. Alan Stall, Juntao Liu, and Dan Ng for help with flow cytometry, and Dr. Paul Rothman for critically reading the manuscript. M.D. is a fellow of the Howard Hughes Medical Institute. S.P.G. is a Howard Hughes Medical Institute investigator.
Footnotes
Abbreviations: IL-7, interleukin 7; FCS, fetal calf serum; AP, abl-positive cultures; AN, abl-negative cultures; TUNEL, terminal deoxynucleotidyltransferase-mediated UTP nick end labeling; BrdU, 5-bromo-2′-deoxyuridine.
References
- 1.Abelson H T, Rabstein L S. Cancer Res. 1970;30:2213–2222. [PubMed] [Google Scholar]
- 2.Wang J Y. Curr Opin Genet Dev. 1993;3:35–43. doi: 10.1016/s0959-437x(05)80338-7. [DOI] [PubMed] [Google Scholar]
- 3.Laneuville P. Semin Immunol. 1995;7:255–266. doi: 10.1006/smim.1995.0030. [DOI] [PubMed] [Google Scholar]
- 4.Chung S W, Wong P M. Oncogene. 1995;10:1261–1268. [PubMed] [Google Scholar]
- 5.Chen Y Y, Rosenberg N. Proc Natl Acad Sci USA. 1992;89:6683–6687. doi: 10.1073/pnas.89.15.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y Y, Wang L C, Huang M S, Rosenberg N. Genes Dev. 1994;8:688–697. doi: 10.1101/gad.8.6.688. [DOI] [PubMed] [Google Scholar]
- 7.Klug C A, Gerety S J, Shah P C, Chen Y Y, Rice N R, Rosenberg N, Singh H. Genes Dev. 1994;8:678–687. doi: 10.1101/gad.8.6.678. [DOI] [PubMed] [Google Scholar]
- 8.Evans C A, Owen-Lynch P J, Whetton A D, Dive C. Cancer Res. 1993;53:1735–1738. [PubMed] [Google Scholar]
- 9.Kabarowski J H, Allen P B, Wiedemann L M. EMBO J. 1994;13:5887–5895. doi: 10.1002/j.1460-2075.1994.tb06934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Owen P J, Musk P, Evans C A, Whetton A D. J Biol Chem. 1993;268:15696–15703. [PubMed] [Google Scholar]
- 11.Sanchez-Garcia I, Grutz G. Proc Natl Acad Sci USA. 1995;92:5287–5291. doi: 10.1073/pnas.92.12.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chapman R S, Whetton A D, Chresta C M, Dive C. Mol Pharmacol. 1995;48:334–343. [PubMed] [Google Scholar]
- 13.Schwartzberg P L, Stall A M, Hardin J D, Bowdish K S, Humaran T, Boast S, Harbison M L, Robertson E J, Goff S P. Cell. 1991;65:1165–1175. doi: 10.1016/0092-8674(91)90012-n. [DOI] [PubMed] [Google Scholar]
- 14.Tybulewicz V L, Crawford C E, Jackson P K, Bronson R T, Mulligan R C. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 15.Hardin J D, Boast S, Schwartzberg P L, Lee G, Alt F W, Stall A M, Goff S P. Cell Immunol. 1995;165:44–54. doi: 10.1006/cimm.1995.1185. [DOI] [PubMed] [Google Scholar]
- 16.Hardin J D, Boast S, Schwartzberg P L, Lee G, Alt F W, Stall A M, Goff S P. Cell Immunol. 1996;172:100–107. doi: 10.1006/cimm.1996.0220. [DOI] [PubMed] [Google Scholar]
- 17.Sawyers C L, McLaughlin J, Goga A, Havlik M, Witte O. Cell. 1994;77:121–131. doi: 10.1016/0092-8674(94)90240-2. [DOI] [PubMed] [Google Scholar]
- 18.Goga A, Liu X, Hambuch T M, Senechal K, Major E, Berk A J, Witte O N, Sawyers C L. Oncogene. 1995;11:791–799. [PubMed] [Google Scholar]
- 19.Welch P J, Wang J Y. Mol Cell Biol. 1995;15:5542–5551. doi: 10.1128/mcb.15.10.5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosti V, Bergamaschi G, Lucotti C, Danova M, Carlo-Stella C, Locatelli F, Tonon L, Mazzini G, Cazzola M. Blood. 1995;86:3387–3393. [PubMed] [Google Scholar]
- 21.Daniel R, Cai Y, Wong P M, Chung S W. Oncogene. 1995;10:1607–1614. [PubMed] [Google Scholar]
- 22.Hock H, Dorsch M, Diamantstein T, Blankenstein T. J Exp Med. 1991;174:1291–1298. doi: 10.1084/jem.174.6.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kincade P W, Lee G, Sun L, Watanabe T. J Immunol Methods. 1981;42:17–26. doi: 10.1016/0022-1759(81)90220-9. [DOI] [PubMed] [Google Scholar]
- 24.Gulley M L, Ogata L C, Thorson J A, Dailey M O, Kemp J D. J Immunol. 1988;140:3751–3757. [PubMed] [Google Scholar]
- 25.Springer T, Galfre G, Secher D S, Milstein C. Eur J Immunol. 1979;9:301–306. doi: 10.1002/eji.1830090410. [DOI] [PubMed] [Google Scholar]
- 26.Coffman R L, Weissman I L. Nature (London) 1981;289:681–683. doi: 10.1038/289681a0. [DOI] [PubMed] [Google Scholar]
- 27.Whitlock C A, Witte O N. Proc Natl Acad Sci USA. 1982;79:3608–3612. doi: 10.1073/pnas.79.11.3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hardy R R, Kishimoto T, Hayakawa K. Eur J Immunol. 1987;17:1769–1774. doi: 10.1002/eji.1830171214. [DOI] [PubMed] [Google Scholar]
- 29.Dorsch M, Hock H, Diamantstein T. Eur J Immunol. 1994;24:2049–2054. doi: 10.1002/eji.1830240917. [DOI] [PubMed] [Google Scholar]
- 30.Vanderplasschen A, Hanon E, Pastoret P P. Biochemica. 1995;1:21. [Google Scholar]
- 31.Hardy R R, Carmack C E, Shinton S A, Kemp J D, Hayakawa K. J Exp Med. 1991;173:1213–1225. doi: 10.1084/jem.173.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rolink A, Kudo A, Karasuyama H, Kikuchi Y, Melchers F. EMBO J. 1991;10:327–336. doi: 10.1002/j.1460-2075.1991.tb07953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee G, Namen A E, Gillis S, Ellingsworth L R, Kincade P W. J Immunol. 1989;142:3875–3883. [PubMed] [Google Scholar]
- 34.Park L S, Friend D J, Schmierer A E, Dower S K, Namen A E. J Exp Med. 1990;171:1073–1089. doi: 10.1084/jem.171.4.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayashi S, Kunisada T, Ogawa M, Sudo T, Kodama H, Suda T, Nishikawa S, Nishikawa S. J Exp Med. 1990;171:1683–1695. doi: 10.1084/jem.171.5.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yasunaga M, Wang F, Kunisada T, Nishikawa S, Nishikawa S. J Exp Med. 1995;182:315–323. doi: 10.1084/jem.182.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sherwood S W, Schimke R T. Methods Cell Biol. 1995;46:77–97. doi: 10.1016/s0091-679x(08)61925-1. [DOI] [PubMed] [Google Scholar]
- 38.Gavrieli Y, Sherman Y, Ben-Sasson S A. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Griffiths S D, Goodhead D T, Marsden S J, Wright E G, Krajewski S, Reed J C, Korsmeyer S J, Greaves M. J Exp Med. 1994;179:1789–1797. doi: 10.1084/jem.179.6.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartzman R A, Cidlowski J A. Int Arch Allergy Immunol. 1994;105:347–354. doi: 10.1159/000236781. [DOI] [PubMed] [Google Scholar]
- 41.Kincade P W. Proc Natl Acad Sci USA. 1994;91:2888–2889. doi: 10.1073/pnas.91.8.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collins M K, Lopez Rivas A. Trends Biochem Sci. 1993;18:307–309. doi: 10.1016/0968-0004(93)90042-l. [DOI] [PubMed] [Google Scholar]
- 43.Baffy G, Miyashita T, Williamson J R, Reed J C. J Biol Chem. 1993;268:6511–6519. [PubMed] [Google Scholar]
- 44.White E. Genes Dev. 1996;10:1–15. doi: 10.1101/gad.10.1.1. [DOI] [PubMed] [Google Scholar]
- 45.Askew D S, Ashmun R A, Simmons B C, Cleveland J L. Oncogene. 1991;6:1915–1922. [PubMed] [Google Scholar]
- 46.Walker P R, Kwast-Welfeld J, Gourdeau H, Leblanc J, Neugebauer W, Sikorska M. Exp Cell Res. 1993;207:142–151. doi: 10.1006/excr.1993.1173. [DOI] [PubMed] [Google Scholar]
- 47.Yonish-Rouach E, Grunwald D, Wilder S, Kimchi A, May E, Lawrence J J, May P, Oren M. Mol Cell Biol. 1993;13:1415–1423. doi: 10.1128/mcb.13.3.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strasser A. Curr Opin Immunol. 1995;7:228–234. doi: 10.1016/0952-7915(95)80007-7. [DOI] [PubMed] [Google Scholar]