

Abstract
Several nickel(0), palladium(II), and rhodium(I) complexes have been prepared using for the first time the stable bis(diisopropylamino)cyclopropenylidene (BAC). Based on single crystal X-ray diffraction studies and spectroscopic data, the structural and electronic properties of these complexes are discussed. Moreover, their similarities and differences with the analogous NHC complexes are emphasized.
Keywords: Cyclopropenylidene, Stable Carbenes, Carbene transition metal complexes, Rhodium, Palladium, Nickel
1. Introduction
Over the years the success of homogeneous catalysis can be attributed largely to the development of a diverse range of ligand frameworks that have been used to tune the behavior of the various systems. In many cases the use of σ-donors as ancillary ligands are required to achieve high efficiency. Spectacular results in this area have been reported using cyclic diaminocarbenes (NHCs) [1] (Fig. 1). It is noteworthy that although NHC-transition metal complexes have been known since the sixties [2], the recent developments in their application as scaffolds in catalysis have only been made possible because of the availability of bottle-able NHCs [3]. Although it is possible to cursorily tune the structure of NHCs, any diversity is still far from matching their phosphorus-based counterparts. Recently, our group has reported the synthesis of two new families of stable carbenes, namely cyclic (alkyl)(amino)carbenes (CAACs) [4], and bis(amino)cyclopropenylidenes (BACs) [5,6]. It has already been shown that CAACs are excellent ligands for transition metal centers, and the catalytic activity of the corresponding complexes can even be superior to that of NHC analogues. Isolated cyclopropenylidenes have never been used to prepare transition metal complexes. However, it should be noted that a few cyclopropenylidene complexes have been prepared via oxidative addition using a carbene precursor [7]. With few exceptions [7b–d,f,h,i] the three-membered ring was substituted by aryl groups. Interestingly, recent works have shown that aryl-substituted cyclopropenylidene palladium complexes are active catalysts for Heck and Suzuki C-C coupling reactions, as well as for Buchwald-Hartwig aryl aminations [7j-1].
Figure 1.
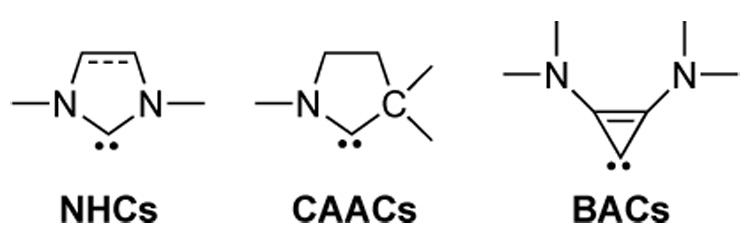
Schematic representation of NHCs, CAACs, and BACs.
From this analysis, it is clear that due to their stability as free species, bis(amino)cyclopropenylidenes (BACs) should give increased flexibility in the synthesis of complexes otherwise inaccessible by existing methodologies. Moreover, due to the presence of the strong π-donor amino groups, BACs should act as strong σ-donor ligands. Here we report the first syntheses of transition metal complexes using stable BACs as ligands.
2. Results and Discussion
First, it was of interest to compare the donor properties of bis(diisopropylamino)cyclopropenylidene 1 with other L ligands. The carbonyl stretching frequencies of cis-[RhCl(CO)2(L)] complexes are recognized as an excellent measure of the σ-donor and π-acceptor properties of ligands L [8]. When a benzene solution of free carbene 1 was reacted with half an equivalent of [RhCl(CO)2]2 a bright red solution was obtained (Scheme 1). The 13C NMR spectrum of the resulting complex 2 revealed a doublet at 141 ppm (JRh-C = 44 Hz). The coupling constant is similar to those observed for other Rh-carbene complexes, and the chemical shift agrees well with the literature, the carbene signal being at half way between that for free 1 and the cyclopropenium salt precursor. The infrared carbonyl stretching frequencies of 2 were observed at v = 2070 and 1992 cm−1 in methylene chloride (vavg = 2031 cm−1). These values indicate that the donor power of BAC 1 is slightly superior to that of unsaturated NHCs (about 2041 cm−1) and even saturated NHCs (about 2038 cm−1) [8].
Scheme 1.
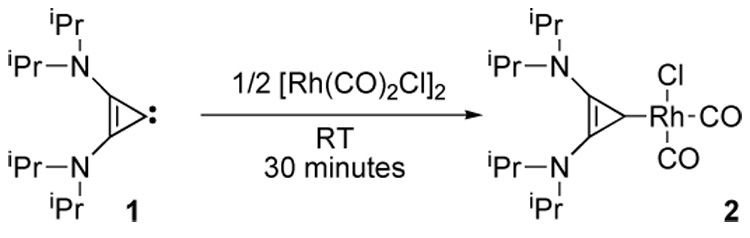
Preparation of the Rh(CO)2(L)Cl complex 2.
Another classical rhodium complex precursor is the [Rh(COD)Cl]2, which usually reacts with an L ligand to afford the monomeric Rh(COD)(L)Cl complex. By combining a benzene solution of free carbene 1 with half an equivalent of [Rh(COD)Cl]2, a new product crystallized readily out of benzene (Scheme 2). X-ray analysis of these crystals demonstrated the unexpected structure 3 shown in figure 2. It is a cationic rhodium complex, featuring two cyclopropenylidene ligands, and Rh(COD)Cl2− as a counter anion. Although a few examples of cationic Rh(L)2(NHC)2 are known [9], a search in the Cambridge data base reveals that the only analog containing both a Rh(COD)(L)(carbene) cation and Rh(COD)Cl2 anion is restricted to chelating pyridine functionalized NHC’s [10]. We believe that the formation of 3 is a good indication that metals are able to accommodate several BACs, thanks to their restricted steric bulk.
Scheme 2.
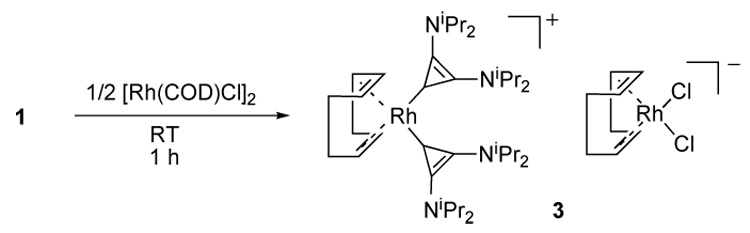
Unexpected formation of complex 3.
Figure 2.
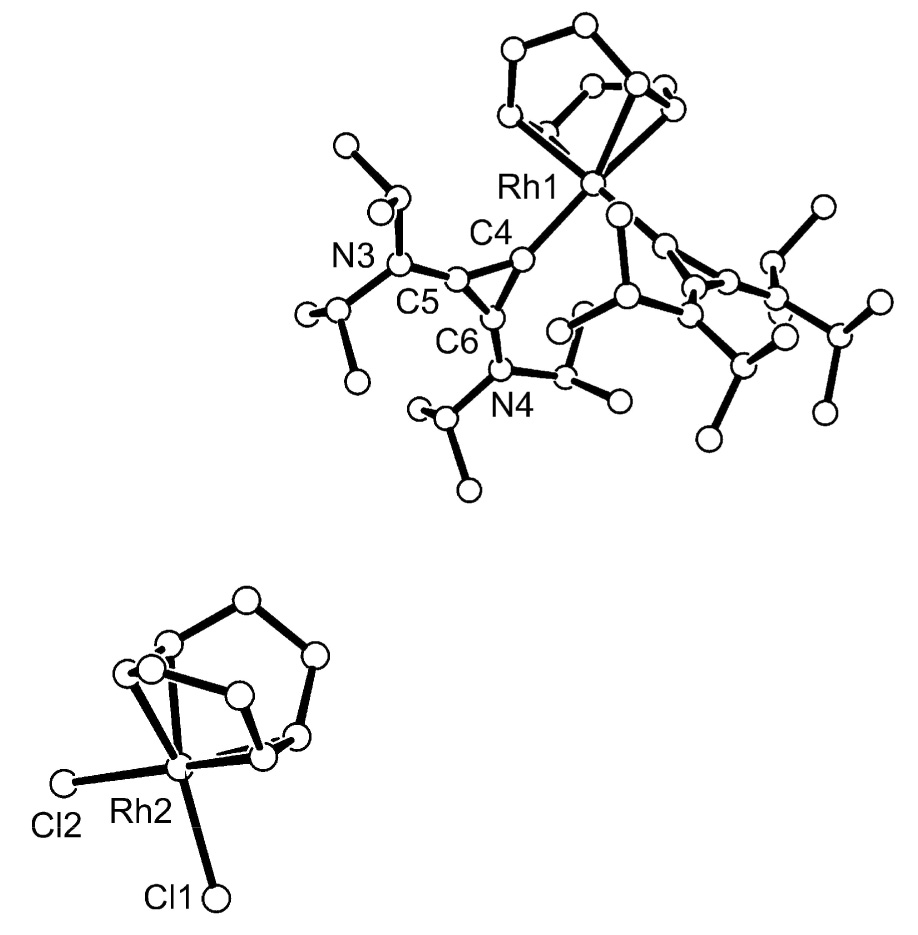
Molecular view of the crystal structure of 3. Selected bond lengths [Å] and angles [°]: Rh(1)-C(4) 2.028(6), C(4)-C(5) 1.380(9), C(4)-C(6) 1.389(9) C(5)-C(6) 1.368(9), N(4)-C(6) 1.324(8), N(3)-C(5) 1.342(8); C(4)-Rh(1)-C(1) 91.8(2), C(6)-C(5)-C(4) 60.8(5), C(5)-C(4)-C(6) 59.2(4), C(5)-C(6)-C(4) 60.0(5).
Since BAC 1 appeared to be a strong σ-donor, standard L ligand exchange reactions should allow for the synthesis of a variety of complexes hardly available without the free ligand. To test this hypothesis we first studied the reaction of 1 with the Wilkinson’s catalyst (Scheme 3). Monitoring the reaction at room temperature by 31P NMR spectroscopy, we observed the primary formation of complex 4a, which gave a pair of doublets of doublets at 53 ppm (1JP-Rh = 206.6 Hz, JP-P = 37.6 Hz) and 37 ppm (JP-Rh = 123.6 Hz, JP-P = 37.6 Hz) indicative of a cis-arrangement of the phosphines. Upon heating for 2 hours at 80°C, a new doublet in the 31P NMR spectrum appears at 33 ppm (JP-Rh = 156.7 Hz) suggesting the formation of the trans-isomer 4b. After work up, the thermodynamic complex 4b was isolated as orange crystals in 91% yield, and unambiguously characterized by single crystal X-ray analysis (Fig. 3). The rhodium-carbene bond length of 1.943 Å is shorter than that for known analogous NHC complexes, which range from 2.053 to 1.987 Å [11].
Scheme 3.
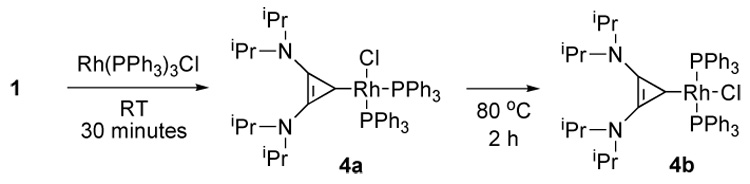
Reaction of carbene 1 with the Wilkinson’s catalyst.
Figure 3.
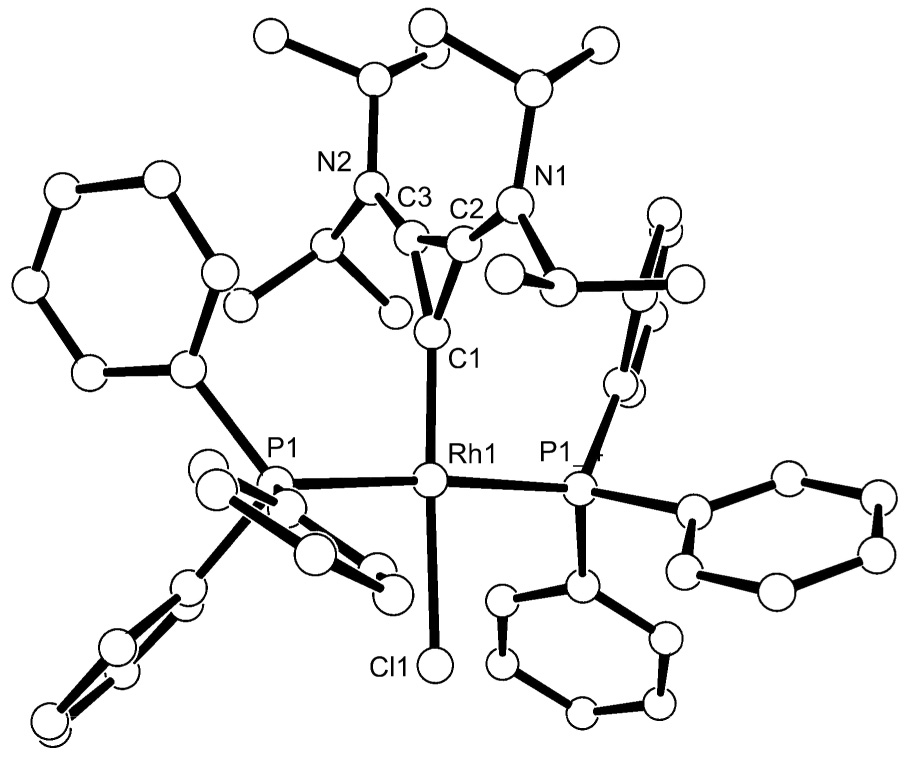
Molecular view of the crystal structure of 4b. Selected bond lengths [Å] and angles [°]: Rh(1)-C(1) 1.943(10), Rh(1)-P(1) 2.294(2), Rh(1)-Cl(1) 2.411(3), C(1)-C(2) 1.352(16), C(1)-C(3) 1.390(15), C(2)-C(3) 1.378(17), N(2)-C(3) 1.367(14), N(1)-C(2) 1.324(16); C(1)-Rh(1)-P(1)#1 92.21(6), C(1)-Rh(1)-P(1) 92.21(6), P(1)#1-Rh(1)-P(1) 174.58(11), C(1)-Rh(1)-Cl(1) 175.2(3), P(1)#1-Rh(1)-Cl(1) 87.66(6), P(1)-Rh(1)-Cl(1) 87.66(6), (2)-C(1)-C(3) 60.3(8), C(2)-C(3)-C(1) 58.5(8).
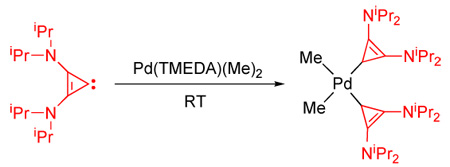
We then investigated the facility with which the free BAC 1 could displace common neutral bidentate ligands. We were pleased to find that two equivalents of free carbene 1 reacted with Pd(TMEDA)(Me)2 [12] in a toluene solution within two hours at room temperature. Colorless crystals of 5 suitable for a single crystal X-ray diffraction study were obtained by slow diffusion of hexanes into a saturated toluene solution cooled to - 10°C (89% yield) (Scheme 4). The metal center is in a distorted square planar environment, and the three-membered rings are tilted by 63° (torsion angle Cmethyl-Pd-C1–C2) with respect to the square plane (Fig. 4). The two palladium-carbene bond lengths are equivalent within experimental error (average 2.027 Å), and are shorter than those observed by Douthwaite for analogous complexes bearing di-NHC [13] and NHC-phosphine [14] bidentate ligands (2.07–2.08 Å).
Scheme 4.
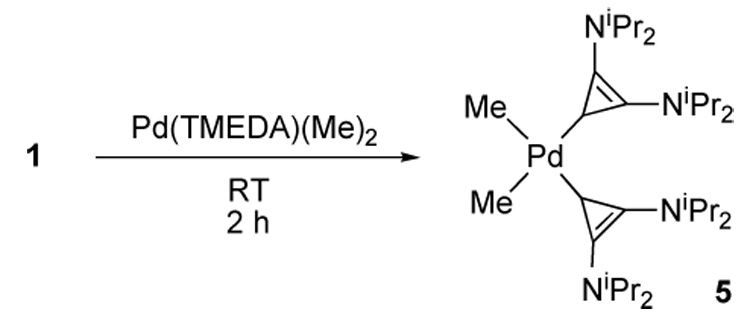
Synthesis of Palladium(II) complex 5.
Figure 4.
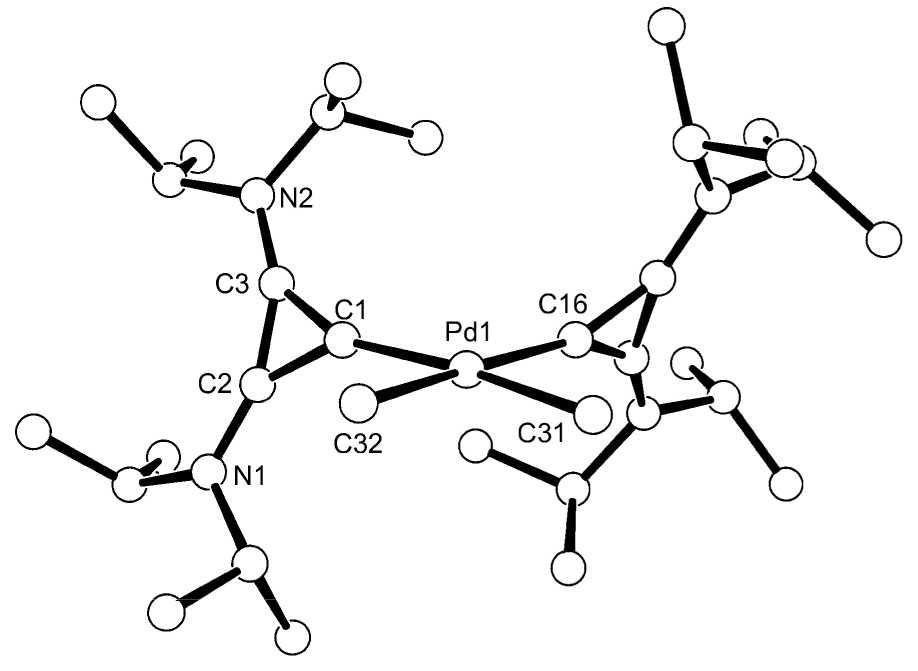
Molecular view of the crystal structure of 5. Selected bond lengths [Å] and angles [°]: Pd(1)-C(1) 2.0242(13), Pd(1)-C(16) 2.0306(12), Pd(1)-C(31) 2.0904(14), Pd(1)-C(32) 2.0913(13), N(1)-C(2) 1.3311(18), N(2)-C(3) 1.3357(18), C(1)-C(2) 1.3903(19), C(1)-C(3) 1.3923(18), C(2)-C(3) 1.3708(19); C(1)-Pd(1)-C(16) 97.42(5), C(1)-Pd(1)-C(31) 173.64(5), C(16)-Pd(1)-C(31) 88.05(6), C(1)-Pd(1)-C(32) 88.61(6), C(16)-Pd(1)-C(32) 173.88(6), C(31)-Pd(1)-C(32) 86.00(6), C(2)-C(1)-C(3) 59.03(9), C(2)-C(3)-C(1) 60.42(10), C(3)-C(2)-C(1) 60.56(10).
Lastly, metal(0) complexes are hardly available without the free carbene, and thus it was of interest to react BAC 1 with bis(1,5-cyclooctadiene)nickel(0) (Scheme 5). The exchange reaction occurred readily in benzene within 10 minutes at room temperature, and complex 6 was isolated as an orange solid in 86% yield. Both 13C and 1H NMR reveal a single set of signals and integration consistent with coordination of two carbene ligands with a single chelating COD. This structure is unknown for the corresponding NHC complexes wherein a bridging COD connects two Ni(iPr2Im)2 fragments [15], and stands in contrast to the homoleptic Ni(IMes)2 product formed via the analogous synthetic route [16]. We believe that the minimal steric bulk of BAC 1 is responsible for the chelation of COD to form the monomeric complex 6.
Scheme 5.
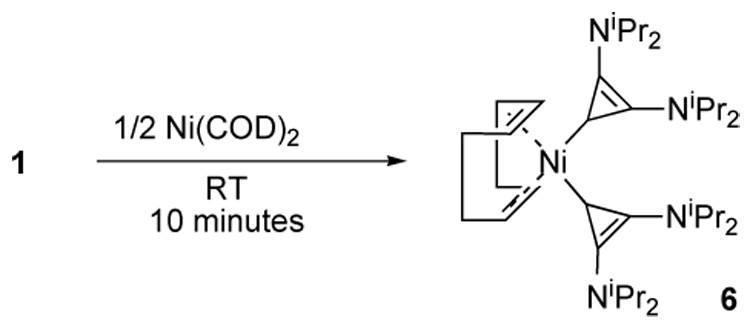
Synthesis of Nickel(0) complex 6.
3. Conclusion
From the infra-red carbonyl stretching frequencies of cis-[RhCl(CO)2(BAC)] complex, it appears that the stable bis(diisopropylamino)cyclopropenylidene is a strong σ-donor ligand. It promotes the cleavage [Rh(COD)Cl]2 but in contrast to NHCs it leads to a Rh(COD)(BAC)2 cationic complex, suggesting that metals can easily accommodate several BAC ligands, because of their unique steric properties. BAC is able to substitute L ligands such as triphenylphosphine, but also bidentate ligands such as TMEDA and COD. The catalytic activity of these and other BAC complexes is under active investigation.
4. Experimental
General
All manipulations were performed under an inert atmosphere of argon using standard Schlenk techniques or using an MBraun glovebox. Dry, oxygen-free solvents were employed. 1H, 13C, and 31P NMR spectra were recorded on Bruker AC200, WM250, Avance 300, Varian Inova 300 or Inova 500 spectrometers. 1H and 13C chemical shifts are reported in ppm relative to Me4Si as external standard, and 31P chemical shifts are reported in ppm relative to H3PO4 as external standard.
4.1. [Bis(diisopropylamino)cyclopropenylidene]cis-dicarbonyl-chlororhodium(I) complex 2
To a solution of cyclopropenylidene 1 (224 mg, 0.90 mmol) in C6D6 (0.5 mL) was added (chloro)(dicarbonyl)rhodium(I) dimer (176 mg, 0.45 mmol). A bright red solution was formed instantaneously, which was stirred at room temperature for 30 minutes. The solvent was removed in vacuo and complex 2 was isolated as a red, microcrystalline solid. Yield: 295 mg (0.69 mmol, 74 %); m.p. 9 5°C, dec.; 1H NMR (300 MHz, C6D6) δ = 3.82-3.60 (broad m, 4 H), 1.49-1.04 (broad d, 24 H); 13C{1H} NMR (75 MHz, C6D6) δ = 187.2 (d, 1JRh-C = 54.3 Hz, CO), 184.3 (d, 1JRh-C = 72.8 Hz, CO), 149.2 (Cring), 141.2 (d, 1JRh-C = 43.9 Hz, Ccarb), 53.34 (broad, CH), 48.64 (broad, CH), 21.83 (broad, CH3); IR (CH2Cl2): v [cm−1] 2070 (s) 1992(s).
4.2 Di[bis(diisopropylamino)cyclopropenylidene](COD)rhodium(I)] [dichloro-(COD) rhodium(III)] 3
To a solution of cyclopropenylidene 1 (52 mg, 0.21 mmol) in C6D6 (0.5 mL), (chloro)(cyclooctadiene)rhodium(I) dimer (74 mg, 0.10 mmol) was added leading to a dark orange solution. Blocky orange crystals of complex 3 were formed upon standing at room temperature overnight. Yield: 103 mg, (0.21 mmol, 81%). 1H NMR (300 MHz, C6D6) δ = 4.20 (broad, 8 H), 4.02 (broad, 8 H), 2.35 (broad, 4 H), 2.15 (broad, 4 H), 2.00 (d, 4 H, J = 8.0 Hz), 1.60 (d, 4 H, J = 7.8 Hz), 1.21 (broad, 48 H); 13C{1H} NMR (75 MHz, C6D6) δ = 149.8 (Cring), 149.1 (d, 1JRh-C = 54.0 Hz, Ccarb), 85.9 (broad, CH), 49.3 (broad, CHCH3), 31.0 (broad, CH2), 21.6 (broad, CHCH3).
4.3. [Bis(diisopropylamino)cyclopropenylidene)]bis(triphenylphosphine)(chloro)-rhodium(I) complexes 4
Cyclopropenylidene 1 (251 mg, 1.00 mmol) was combined with Wilkinson’s catalyst (924 mg, 1.00 mmol), and the mixture dissolved in freshly distilled toluene (20 mL) with stirring at room temperature for one hour. Monitoring this reaction by multinuclear NMR spectroscopy allowed for the characterization of the cis-isomer 4a. 31P{1H} NMR (121 MHz, C6D6) δ = 53.4 (dd, 1JP-Rh = 206.6 Hz, 2JP-P = 37.6 Hz), 37.7 (dd, 1JP-Rh = 123.6 Hz, 2JP-P = 37.6 Hz). 1H NMR (300 MHz, C6D6) δ = 8.09 (m, 5 H), 7.88 (m, 5 H), 7.09 (m, 10 H), 7.00 (m, 10H), 4.04 (m, 4 H), 1.43-0.96 (m, 24 H); 13C{1H} NMR (75 MHz, C6D6) δ = 147.5 (Cring), 138.2 (d, 1JRh-C = 33.4 Hz, Ccarb), 136.0 (d, J = 11.3 Hz), 134.6 (d, J = 12.3 Hz), 128.0, 126.9-126.6 (m), 49.4, 21.8. Then the toluene solution was heated at 80 °C for 2 h. The solvent was removed in vacuo to afford a brown oil. This material was washed with hexanes leading to the trans-isomer 4b as an off-white microcrystalline solid. Yield: 816 mg (0.91 mmol, 91 % yield). Single crystals were obtained from a saturated benzene solution at room temperature; mp 218 °C, dec; 31P{1H} NMR (121 MHz, C6D6) δ = 33.0 (d, J = 156.7 Hz); ); 1H NMR (300 MHz, C6D6) δ = 7.74-6.98 (m, 30 H), 4.34 (m, 4 H), 1.34-0.84 (m, 24 H); 13C{1H} NMR (75 MHz, C6D6) δ = 148.9 (Cring), 139.8 (d, 1JRh-C = 39.3 Hz, Ccarb), 135.4 (m), 132.1 (d, J = 9.6 Hz), 131.4, 127.8, 48.8, 22.3.
4.4. Cis-Di[bis(diisopropylamino)cyclopropenylidene]-di(methyl)palladium(II) complex 5
In the glovebox, cyclopropenylidene 1 (767 mg, 3.23 mmol) was added to a solution of Pd(TMEDA)(Me)2 (386 mg, 1.53 mmol) in toluene (10mL) and stirred at room temperature for 2 hours. After removal of the volatiles with gentle heating under vacuum, the resulting yellow, sticky foam was washed with hexanes giving a fine grey powder. The complex recrystallized out of a minimum of warm toluene, layered with hexanes, and cooled to −40 °C overnight led to complex 5, which was isolated as colorless, blocky crystals. Yield: 834 mg (1.37 mmol, 89 %); m.p. 107°C, dec; 1H NMR (300 MHz, C6D6) δ = 4.11 (broad s, 8 H); 1.29 (d, J = 6.48 Hz, 48 H); 0.57 (s, 6 H); 13C{1H} NMR (75 MHz, C6D6) δ = 165.5 (Ccarb); 148.8 (Cring); 48.9 (CHCH3); 21.7 (CHCH3); −3.8 (CH3); TOF HRMS calculated for C31H59N4Pd [M-CH3]+: m/z 593.3774; found 593.3768.
4.5. Di[bis(diisopropylamino)cyclopropenylidene)](COD)nickel(0) complex 6
Cyclopropenylidene 1 (115 mg, 0.458 mmol) was dissolved in C6D6 (0.5 mL) and the solution added to bis(COD)nickel(0) (63 mg, 0.229 mmol) at room temperature. The solution immediately changed color from yellow to red. After stirring for 1 h, the volatiles were removed in vacuo to give complex 6 as an orange solid. Yield 126 mg (0.197 mmol, 86 %); m.p. 121 °C, dec; 1H NMR (300 MHz, C6D6) δ = 4.41 (broad s, 4 H), 3.95 (sept, 8 H), 2.42 (broad s, 8 H), 1.27 (d, 48 H); 13C{1H} NMR (75 MHz, C6D6) δ = 176.8 (Ccarb), 149.2 (Cring), 92.1, 49.5, 33.7, 22.8.
4.6. Crystal structure determination of complexes 3, 4b and 5
The Bruker X8-APEX X-ray diffraction instrument with Mo-radiation was used for data collection. All data frames were collected at low temperatures (T = 100 K) using an ω, φ-scan mode (0.5° ω-scan width, hemisphere of reflections) and integrated using a Bruker SAINTPLUS software package. The intensity data were corrected for Lorentzian polarization. Absorption corrections were performed using the SADABS program. The SIR97 was used for direct methods of phase determination, and Bruker SHELXTL software package for structure refinement and difference Fourier maps. Atomic coordinates, isotropic and anisotropic displacement parameters of all the non-hydrogen atoms of compounds were refined by means of a full matrix least-squares procedure on F2. All H-atoms were included in the refinement in calculated positions riding on the C atoms. Drawings of molecules were performed using Ortep 3. Crystal and structure parameters of 3: size 0.22 × 0.13 × 0.10 mm3, monoclinic, space group P 2(1)/n, a = 11.8164(15) Å, b = 22.759(3) Å, c = 20.536(3) Å, α= γ =90.0°, β = 90.907(2)°, V = 5522.0(12) Å3, ρcalcd = 1.303 g/cm3, Mo-radiation (λ = 0.71073 Å), T = 100(2) K, reflections collected = 24998, independent reflections = 5930 (Rint = 0.0860), absorption coefficient μ = 0.732 mm−1; max/min transmission = 0.9304 and 0.8556, 641 parameters were refined and converged at R1 = 0.0449, wR2 = 0.0900, with intensity I>2σ(I). Crystal and structure parameters of 4b: size 0.36 × 0.13 × 0.09 mm3, monoclinic, space group P 2(1)/m, a = 11.379(3) Å, b = 23.388(7) Å, c = 11.507(4) Å, α = γ = 90.0° β = 104.245(4)°, V = 2968.0(16) Å3, ρcalcd = 1.006 g/cm3, Mo-radiation (λ = 0.71073 Å), T = 100(2) K, reflections collected = 16677, independent reflections = 4385 (Rint = 0.0970), absorption coefficient μ = 0.414 mm−1 ; max/min transmission = 0.9637 and 0.8651, 278 parameters were refined and converged at R1 = 0.0891, wR2 = 0.1886, with intensity I>2σ(I). Crystal and structure parameters of 5: size 0.34 × 0.21 × 0.10 mm3, monoclinic, space group P 2(1)/c, a = 14.7185(5) Å, b = 14.8792(5) Å, c = 19.3392(7) Å, α γ = 90.0°, β = 110.208(2)°, V = 3974.6(2) Å3, ρcalcd = 1.018 g/cm3, Mo-radiation (λ = 0.71073 Å), T = 100(2) K, reflections collected = 76871, independent reflections = 13230 (Rint = 0.0246), absorption coefficient μ = 0.488 mm−1; max/min transmission = 0.8557 and 0.7961 352 parameters were refined and converged at R1 = 0.0286, wR2 = 0.0761, with intensity I>2σ(I). Structural data for compounds 3, 4b and 5 have been deposited in the Cambridge Crystallographic Data Center under CCDC 662010–662012, and can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html.
Acknowledgements
Thanks are due to the NIH (R01 GM 68825) and RHODIA for financial support of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Synopsis For the first time, the stable bis(diisopropylamino)cyclopropenylidene is used to prepare transition metal complexes via ligand exchange.
References
- 1.For reviews on stable carbenes as ligands for transition metal catalysts, see: Diez-Gonzalez S, Nolan SP. Coord. Chem. Rev. 2007;251:874. Marion N, Diez-Gonzalez A, Nolan SP. Angew. Chem. Int. Ed. 2007;46:2988. doi: 10.1002/anie.200603380. Pugh D, Danopoulos AA. Coord. Chem. Rev. 2007;251:610. Nolan SP. N-Heterocyclic Carbenes in Synthesis. Wiley-VCH; 2006. Glorius F. N-Heterocyclic Carbenes in Transition Metal Catalysis (Topics in Organometallic Chemistry) Springer Verlag: 2006. Scott NM, Nolan SP. Eur. J. Inorg. Chem. 2005:1815. Crabtree RH. J. Organometal. Chem. 2005;690:5451. Peris E, Crabtree RH. Coord. Chem. Rev. 2004;248:2239. Crudden CM, Allen DP. Coord. Chem. Rev. 2004;248:2247. César V, Bellemin-Laponnaz S, Gade LH. Chem. Soc Rev. 2004;33:619. doi: 10.1039/b406802p. Herrmann WA. Angew. Chem. Int. Ed. 2002;41:1290. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y.
- 2.a) Öfele K. J. Organometal. Chem. 1968;72:42. [Google Scholar]; b) Cardin DJ, Cetinkaya B, Lappert MF. Chem. Rev. 1972;72:545. [Google Scholar]
- 3.For reviews on stable singlet carbenes: Hahn FE. Angew. Chem. Int. Ed. 2006;45:1348. doi: 10.1002/anie.200503858. Kirmse W. Angew. Chem. Int. Ed. 2004;43:1767. doi: 10.1002/anie.200301729. Alder RW, Blake ME, Chaker ME, Harvey JN, Paolini F, Schütz J. Angew. Chem. Int. Ed. 2004;43:5896. doi: 10.1002/anie.200400654. Canac Y, Soleilhavoup M, Conejero S, Bertrand G. J. Organomet. Chem. 2004;689:3857. Bourissou D, Guerret O, Gabbaï FP, Bertrand G. Chem. Rev. 2000;100:39. doi: 10.1021/cr940472u.
- 4.a) Lavallo V, Mafhouz J, Canac Y, Donnadieu B, Schoeller WW, Bertrand G. J. Am. Chem. Soc. 2004;126:8670. doi: 10.1021/ja047503f. [DOI] [PubMed] [Google Scholar]; b) Lavallo V, Canac Y, Prasang C, Donnadieu B, Bertrand G, Angew G. Chem. Int. Ed. 2005;44:5705. doi: 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lavallo V, Canac Y, DeHope c, Donnadieu B, Bertrand G. Angew. Chem. Int. Ed. 2005;44:7236. doi: 10.1002/anie.200502566. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lavallo V, Canac Y, Donnadieu B, Schoeller WW, Bertrand G. Angew. Chem. Int. Ed. 2006;45:3488. doi: 10.1002/anie.200600987. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Jazzar R, Dewhurst RD, Bourg JB, Donnadieu B, Canac Y, Bertrand G. Angew. Chem. 2007;46:2899. doi: 10.1002/anie.200605083. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Frey GD, Lavallo V, Donnadieu B, Schoeller WW, Bertrand G. Science. 2007;316:439. doi: 10.1126/science.1141474. [DOI] [PubMed] [Google Scholar]; g) Anderson DR, Lavallo V, O’Leary DJ, Bertrand G, Grubbs RH. Angew. Chem. Int. Ed. 2007;46:7262. doi: 10.1002/anie.200702085. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Lavallo V, Frey GD, Kouzar S, Donnadieu B, Bertrand G. Proc. Natl. Acad. Sci. USA. 2007;104:13569. doi: 10.1073/pnas.0705809104. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Masuda JD, Schoeller WW, Donnadieu B, Bertrand G. Angew. Chem. Int. Ed. 2007;46:7052. doi: 10.1002/anie.200703055. [DOI] [PubMed] [Google Scholar]
- 5.a) Lavallo V, Canac Y, Donnadieu B, Schoeller W, Bertrand G. Science. 2006;312:722. doi: 10.1126/science.1126675. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lavallo V, Ishida Y, Donnadieu B, Bertrand G. Angew. Chem. Int. Ed. 2006;45:6652. doi: 10.1002/anie.200602701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.A second example of stable cyclopropenylidene has recently been reported: Holschumacher D, Hrib CG, Jones PG, Tamm M. Chem. Commun. 2007:3661. doi: 10.1039/b706708a.
- 7.a) Öfele K. Angew. Chem., Int. Ed. Engl. 1968;7:950. [Google Scholar]; b) Weiss R, Priesner C. Angew. Chem., Int. Ed. Engl. 1978;17:457. [Google Scholar]; c) Konishi H, Matsumoto S, Kamatori Y, Ogoshi H, Yoshida Z. Chem. Let. 1978:241. [Google Scholar]; d) Wilson RD, Kamitori Y, Ogoshi H, Yoshida Z, Ibers JA. J. Organomet. Chem. 1979;173:199. [Google Scholar]; e) Kawada Y, Jones WM. J. Organomet. Chem. 1980;192:87. [Google Scholar]; f) Yoshida Z. Pure Appl. Chem. 1982;54:1059. [Google Scholar]; g) Miki S, Ohno T, Iwasaki H, Yoshida Z. J. Phys. Org. Chem. 1988;1:333. [Google Scholar]; h) Miki S, Ohno T, Iwasaki H, Maeda y, Yoshida Z. Tetrahedron. 1988;44:55. [Google Scholar]; i) Tamm M, Grzegorzewski A, Hahn FE. J. Organometal. Chem. 1995;501:309. [Google Scholar]; j) Wass D, Haddow M, Hey T, Orpen A, Russell C, Shishkov I, Wingad R, Green M. Chem. Comm. 2007:2704. doi: 10.1039/b702827j. [DOI] [PubMed] [Google Scholar]; k) Herrmann WA, Öfele K, Taubmann C, Herdtweck E, Hoffmann S. J. Organomet. Chem. 2007;692:3846. [Google Scholar]; l) Wass D, Hey T, Rodriguez-Castro J, Russell C, Shishkov I, Wingad R, Green M. Organometallics. 2007;26:4702. [Google Scholar]
- 8.a) Herrmann WA, Schütz J, Frey GD, Herdtweck E. Organometallics. 2006;25:2437. [Google Scholar]; b) Mayr M, Wurst K, Ongania KH, Buchmeiser M. M. Chem. Eur. J. 2004;10:1256. doi: 10.1002/chem.200305437. [DOI] [PubMed] [Google Scholar]; c) Herrmann WA, Öfele K, Preysing Dv, Herdtweck E. E. J. Organomet. Chem. 2003;684:235. [Google Scholar]
- 9.See for examples: Herrmann WA, Fischer J, Öfele K, Artus GRJ. J. Organomet. Chem. 2001;530:259. Huang J, Stevens ED, Nolan SP. Organometallics. 2000;19:1194. Park KH, Kim SY, Son SU, Chung YK. Eur. J. Org. Chem. 2003:4341. Schütz J, Herrmann WA. J. Organomet. Chem. 2004;689:2995. Mata JA, Peris E, Incarvito C, Crabtree RH. R. H. J. Chem. Soc., Chem. Commun. 2003:184. doi: 10.1039/b210726k. Alcarazo M, Roseblade SJ, Cowley AR, Fernandez R, Brown JM, Lassaletta JM. J. M. J.Am. Chem. Soc. 2005;127:3290. doi: 10.1021/ja0423769.
- 10.Stylianides N, Danopoulos AA, Tsoureas N. J. Organomet. Chem. 2005;690:5948. [Google Scholar]
- 11.a) Grasa G, Moore Z, Martin K, Stevens E, Nolan S, Paquet V, Lebel H. J. Organomet. Chem. 2002;658:126. [Google Scholar]; b) Allen D, Crudden C, Calhoun L, Wang R, Decken A. J. Organomet. Chem. 2005;690:5736. [Google Scholar]; c) Herrmann W, Frey GD, Herdtweck E, Steinbeck M. Adv. Synth. Catal. 2007;349:1677. [Google Scholar]
- 12.de Graaf W, Boersma J, Smeets WJJ, Spek AL, Van Koten G. Organometallics. 1989;12:2907. [Google Scholar]
- 13.Douthwaite RE, Green MLH, Silcock PJ, Gomes PT. J. Chem. Soc., Dalton Trans. 2002:1386. [Google Scholar]
- 14.Tsoureas N, Danopoulos AA, Tulloch AAD, Light ME. Organometallics. 2003;22:4750. [Google Scholar]
- 15.a) Schaub T, Radius U. Chem. Eur. J. 2005;11:5024. doi: 10.1002/chem.200500231. [DOI] [PubMed] [Google Scholar]; b) Schaub T, Backes M, Radius U. Organometallics. 2006;25:4196. [Google Scholar]
- 16.a) Arduengo AJ, Gamper SF, Calabrese JC, Davidson F. J. Am. Chem. Soc. 1994;116:4391. [Google Scholar]; b) Arnold PL, Cloke GN, Geldbach T, Hitchcock PB. Organometallics. 1999;18:3228. [Google Scholar]; c) Clement ND, Cavall KJ, Jones C, Elsevier CJ. Angew. Chem. Int. Ed. 2004;43:1277. doi: 10.1002/anie.200353409. [DOI] [PubMed] [Google Scholar]; d) Caddick S, Cloke FGN, Hitchcock PB, Lewis AKdeK. Angew. Chem. Int. Ed. 2004;43:5824. doi: 10.1002/anie.200460955. [DOI] [PubMed] [Google Scholar]