

Abstract
Background
Cocaine is a widely abused psychostimulant that has both rewarding and aversive properties. While the mechanisms underlying cocaine’s rewarding effects have been studied extensively, less attention has been paid to the unpleasant behavioral states induced by cocaine, such as anxiety.
Methods
In this study we evaluated the performance of dopamine β-hydroxylase knockout (Dbh −/−) mice, which lack norepinephrine (NE), in the elevated plus maze (EPM) to examine the contribution of noradrenergic signaling to cocaine-induced anxiety.
Results
We found that cocaine dose-dependently increased anxiety-like behavior in control (Dbh +/−) mice, as measured by a decrease in open arm exploration. Dbh −/− mice had normal baseline performance in the EPM, but were completely resistant to the anxiogenic effects of cocaine. Cocaine-induced anxiety was also attenuated in Dbh +/− mice following administration of disulfiram, a DBH inhibitor. In experiments using specific adrenergic antagonists, we found that pretreatment with the β-adrenergic receptor antagonist propranolol blocked cocaine-induced anxiety-like behavior in Dbh +/− and wild-type C57BL6/J mice, while the α1 antagonist prazosin and the α2 antagonist yohimbine had no effect.
Conclusions
These results indicate that noradrenergic signaling via β-adrenergic receptors is required for cocaine-induced anxiety in mice.
Keywords: norepinephrine, dopamine β-hydroxylase, disulfiram, cocaine, elevated plus maze, anxiety
Introduction
Cocaine is a widely abused psychostimulant drug that acts by blocking the plasma membrane transporters for dopamine (DA), norepinephrine (NE), and serotonin. In humans, cocaine use results in a broad spectrum of effects, both subjectively positive (e.g. euphoria, increased energy, enhanced alertness) and negative (e.g. anxiety, paranoia, nausea, hypertension). In addition to its well-documented rewarding and locomotor activating effects in rodents, cocaine also induces anxiety-like behavior that can be reversed by administration of typical anxiolytic drugs, such as diazepam (1–7). Whereas cocaine-induced reward has been studied extensively, less is known about the processes underlying the negative behavioral states associated with acute administration of the drug.
Although DA signaling has been primarily implicated in psychostimulant responses, cocaine also increases extracellular NE levels, and NE transmission has been shown to modulate psychostimulant-induced behaviors and neurochemistry (8–11). Given that NE modulates general stress and anxiety responses (12,13), we surmised that NE might also play a critical role in cocaine-induced anxiogenesis.
Dopamine β-hydroxylase (DBH) is the enzyme that converts DA to NE in the catecholamine biosynthetic pathway, and therefore Dbh knockout (Dbh −/−) mice lack NE completely (14,15). Previously, we have shown that Dbh −/− mice exhibit an increase in striatal high affinity-state DA receptors and a corresponding hypersensitivity to the locomotor activating, rewarding, and aversive effects of cocaine (9). In particular, we observed a novel cocaine-induced place aversion in Dbh −/− mice at a dose of 20 mg/kg, a dose that produces a robust place preference in control animals. An initial goal of the present experiments was to determine whether this place aversion in Dbh −/− mice could be attributed to an increase in cocaine-induced anxiety. Few studies to date have explored the specific pathways involved in this particular drug effect, and none have thoroughly examined the role of NE. Using various pharmacological treatments in control and Dbh −/− mice, we assessed the influence of global NE deficit and DA system hypersensitivity on cocaine-induced anxiety. We then examined which particular subtypes of adrenergic receptors are involved in the expression of this behavior.
Materials and Methods
Animals
Male and female Dbh +/− and −/− mice (aged 2 to 5 months) were individually housed on a reversed light cycle (lights on at 19:00, lights off at 7:00), and were allowed a minimum of two weeks to habituate to the new lighting conditions after moving from normal light cycle (lights on at 7:00, lights off at 19:00). Food and water were available ad libitum throughout the course of the study. Data from male and female mice were combined, since there were no detectable gender differences. Dbh mice were generated as described (15) and maintained on a mixed C57Bl6/J and 129SvEv background. Dbh +/− mice were used as controls, because they have normal brain catecholamine levels and are behaviorally identical to wild-type (Dbh +/+) mice (14–16). Three-month old male and female C57BL6/J mice (Jackson Labs, Bar Harbor, MN) were also used to generalize the findings from these experiments to a different strain of wild-type mouse. Housing, handling, and testing conditions for these animals were identical to those used in experiments with Dbh +/− mice. All animals were treated in accordance with the NIH Intramural Animal Care and Use Program guidelines. The experiments described in this article followed the Emory University Division of Animal Resources’ Guide for the Care and Use of Laboratory Animals and were approved by the Emory IACUC committee.
Behavioral testing
The EPM apparatus consisted of two open arms and two enclosed arms arranged in a plus orientation. The arms were elevated 30 inches above the floor, with each arm projecting 12 inches from the center. Because rodents naturally prefer dark, enclosed compartments, a greater willingness to explore the open, well-lit arms is believed to represent a decrease in the animal’s anxiety. This interpretation has been validated by the efficacy of known anxiolytic and anxiogenic treatments in this paradigm (7,12,17,18).
In all experiments, cocaine was injected 20 minutes prior to behavioral testing as described by Yang and colleagues (3). To begin each test, mice were placed in the EPM facing one of the open arms and allowed to freely explore the apparatus for five minutes, during which time their behavior was videotaped. Videotapes were later scored by an observer who was blind to genotype and treatment group. The measure used for analysis was the percentage of time spent exploring the open arms, which was calculated by dividing the time spent in the open arms by the combined time spent in open and closed arms. Because some drug treatments and genetic manipulations alter overall locomotor activity, it is important to use this percentage measurement as the dependent variable for analysis (17). Entry into an arm of the plus maze was defined as the animal placing all four paws in that particular compartment of the apparatus. All tests were run during the dark cycle, between 14:00 and 18:00. Mice were excluded from data analysis for any of the following reasons: if they jumped or fell off the maze after test had begun, if they had a seizure while on the testing apparatus, or if their open arm time was detected as an outlier using Grubb’s test. Of 253 total mice tested, 10 were excluded from data analysis. Data were analyzed using independent samples t-tests, one-way ANOVA followed by Dunnett’s post-hoc tests, or two-way ANOVA followed by Bonferroni post-hoc tests using Prism 4.0 for Macintosh.
Experiment I: Cocaine dose-response
Dbh +/− and −/− mice (n=8 per group) were injected with 0.9% saline (i.p., 10 ml/kg) or cocaine (5, 10, or 20 mg/kg, i.p. at 10 ml/kg, dissolved in 0.9% saline) 20 minutes prior to behavioral testing. Behavioral testing then proceeded for five minutes, as described above.
Experiment II: NE restoration in Dbh −/− mice
Central NE was restored acutely to Dbh −/− mice (n=6–8 per group) prior to testing on the EPM. L-3,4-dihydroxyphenylserine (DOPS) can be converted to NE by the enzyme aromatic amino acid decarboxylase (AADC), thus bypassing the requirement for DBH in the NE synthesis pathway. However, DOPS administration does not alter DA levels in Dbh −/− mice (15). We administered DOPS in combination with benserazide, an AADC inhibitor that does not cross the blood-brain barrier, thereby confining NE restoration to the central nervous system (19). Dbh −/− mice were injected with DOPS (1 g/kg, s.c. + 0.25 g/kg benserazide, s.c.) or vehicle (distilled water with 2% HCl, 2% NaOH, and 2 mg/kg vitamin C, s.c.) four to six hours prior to injection of cocaine (10 mg/kg, i.p in a volume of 10 ml/kg, dissolved in 0.9% saline) or saline. To habituate mice to s.c. injection of large fluid volume, vehicle injections were given for three days prior to test day. Cocaine injection occurred 20 minutes before introducing mice to the EPM, and behavioral testing proceeded for five minutes, as described above.
Experiment III: DBH inhibition in Dbh +/− mice
DBH enzyme activity was inhibited pharmacologically via acute administration of disulfiram. Disulfiram is a copper-chelating agent that inhibits DBH activity and alters catecholamine tissue content (16,20,21). Mice (n=8 per group) were given three injections, each spaced two hours apart, with either vehicle (0.9% saline) or disulfiram (200 mg/kg, i.p. at 10 ml/kg, sonicated and suspended in 0.9% saline). This dosing regimen is known to decrease NE by ~ 70% in the mouse brain (16). Mice received cocaine (10 mg/kg, i.p. at 10 ml/kg, dissolved in 0.9% saline) or saline two hours after the final pretreatment injection, and behavioral testing took place 20 minutes later, as described above. To habituate the mice to the multiple daily injection regimen and large total injection volumes, they were injected three times per day (spaced two hours apart) with 0.9% saline (10 ml/kg) for three days prior to test day.
Experiment IV: Administration of adrenergic antagonists in Dbh +/− mice
Dbh +/− mice (n=10–17 per group) were pretreated with 0.9% saline (4 ml/kg, i.p.), vehicle (0.9% saline with 1.5% DMSO, 1.5% Cremaphor EL, 10 ml/kg, i.p), the β-adrenergic receptor (β-AR) antagonist propranolol (5 mg/kg, i.p. at 4 ml/kg, dissolved in 0.9% saline), the α1-AR antagonist prazosin (0.5 mg/kg, i.p. at 10 ml/kg, dissolved in vehicle), or the α2-AR antagonist yohimbine (2.5 mg/kg, i.p. at 10 ml/kg, dissolved in distilled water) 10 minutes prior to cocaine injection. Behavioral testing was then performed 20 minutes after cocaine injection (10 mg/kg, i.p. at 10 ml/kg, dissolved in saline). Open arm times for the saline and vehicle groups were compared, and no differences were found; therefore these two groups were combined to form a single “control” group.
Experiment V: β-adrenergic inhibition in C57BL6/J wild type mice
C57BL6/J wild type mice (n=7 per group) were pretreated with 0.9 % saline (10 ml/kg, i.p.) or propranolol (5 mg/kg, i.p. at 10 ml/kg, dissolved in 0.9% saline) 10 minutes prior to injection of saline or cocaine (10 or 20 mg/kg, i.p.). Behavioral testing was performed 20 minutes following cocaine injection, as described above. In preliminary experiments with this mouse strain, we found that the 10 mg/kg dose of cocaine was not sufficient to induce significant cocaine-induced anxiety in our laboratory (data not shown). Therefore, we report data only for the 20 mg/kg dose.
Results
Cocaine-induced anxiety is abolished in Dbh −/− mice
Consistent with our previous results (22), baseline performance on the EPM was similar for Dbh +/− and Dbh −/− mice (Fig. 1). Cocaine treatment dose-dependently decreased percent open arm time in Dbh +/− mice. In contrast, the anxiety behavior of Dbh −/− mice was unaffected by cocaine treatment at any dose (Fig. 1). Two-way ANOVA revealed main effects of dose (F[3,56]=4.391, p=0.0076) and genotype (F[1,56]=19.78, p<0.0001), as well as a dose-genotype interaction (F[3,56]=4.046, p=0.0113). Bonferroni post-hoc analysis indicated a significant decrease in percent open arm time only in Dbh +/− mice treated with 10 mg/kg cocaine (p<0.01) and 20 mg/kg cocaine (p<0.01), when compared to saline treated Dbh +/− animals. Also, Dbh +/−animals showed a lower level of open arm exploration when compared to Dbh −/− mice for doses of 10 mg/kg (p<0.01) and 20 mg/kg cocaine (p<0.001).
Figure 1.
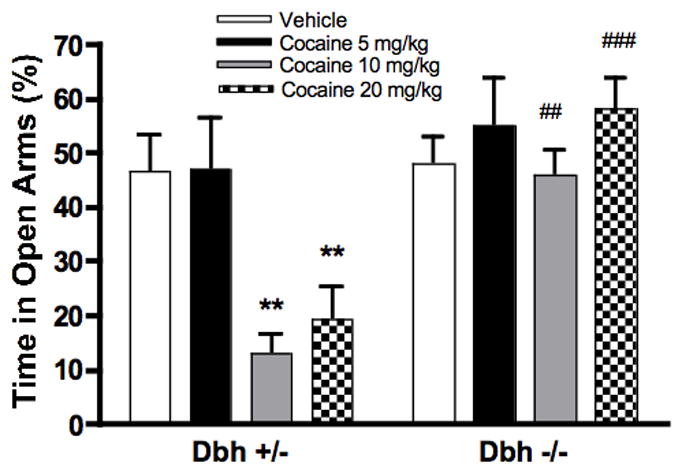
Effects of cocaine on performance in the elevated plus maze in Dbh +/− and Dbh −/− mice. Cocaine was administered to mice 20 minutes prior to the EPM. Shown is percent open arm time during the five minute test. **p<0.01 compared to vehicle control for that genotype. ##p<0.01, ###p<0.001 compared to Dbh +/− mice for that dose (N=8 per group).
Pharmacological restoration of NE alters anxiety behavior in Dbh −/− mice
Dbh −/− mice lack NE from birth and have high tissue DA levels (14), raising the possibility that the failure of cocaine to produce anxiety-like behavior in the knockouts is a result of compensatory mechanisms related either to NE deficiency during postnatal development or to an increase in DA. To test this possibility, we acutely restored central NE to adult Dbh −/− mice without normalizing their DA levels by administering DOPS + benserazide prior to cocaine administration and EPM exposure. Two way ANOVA revealed a main effect of pretreatment (F[1,24]=7.035, p=0.0139, Figure 2). Post-hoc Bonferroni tests indicated that percent open arm time was significantly lower in the DOPS-Cocaine group relative to the Vehicle-Cocaine group (p<0.05), indicating that DOPS treatment restored cocaine-induced anxiety to Dbh −/− mice. However, DOPS treatment also appeared to reduce open arm exploration in saline-treated Dbh −/− mice, and the DOPS-saline group showed an equally low level of open arm exploration as the DOPS-cocaine group. Thus, this particular experiment did not definitively determine whether the increased anxiety in the DOPS-Cocaine animals was due to NE restoration, cocaine, or a combination of both.
Figure 2.
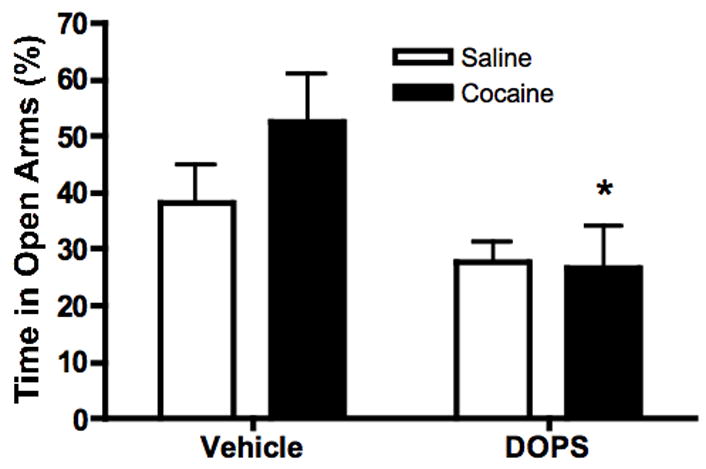
NE replacement alters anxiety behavior in Dbh −/− mice. DOPS + benserazide or vehicle was administered approximately five hours prior to EPM testing, when brain restoration of NE peaks in Dbh −/− mice treated with DOPS. Shown is percent open arm time of Dbh −/− mice given vehicle or cocaine (10 mg/kg, i.p.) 20 minutes before the five minute EPM test. *p<0.05 compared to Vehicle-Cocaine group (N=6–8 per group).
The DBH inhibitor disulfiram abolishes cocaine-induced anxiety in control mice
To determine whether NE depletion confers resistance to cocaine-induced anxiety in normal animals, Dbh +/− mice were pretreated with the DBH inhibitor disulfiram or vehicle prior to cocaine administration and EPM testing. Disulfiram abolished the ability of cocaine to reduce open arm exploration time in Dbh +/− mice, but had no effect in animals treated with saline prior to testing (Figure 3). Two way ANOVA revealed a pretreatment by drug treatment interaction (F[1,28]=5.227, p=0.03). Bonferroni post-hoc tests indicated that the Disulfiram-Cocaine group showed a significantly increased level of open arm exploration relative to the Vehicle-Cocaine group (p<0.05). This suggests that temporary inhibition of NE production decreases cocaine-induced anxiety, phenocopying the behavior we observed in Dbh −/− mice.
Figure 3.
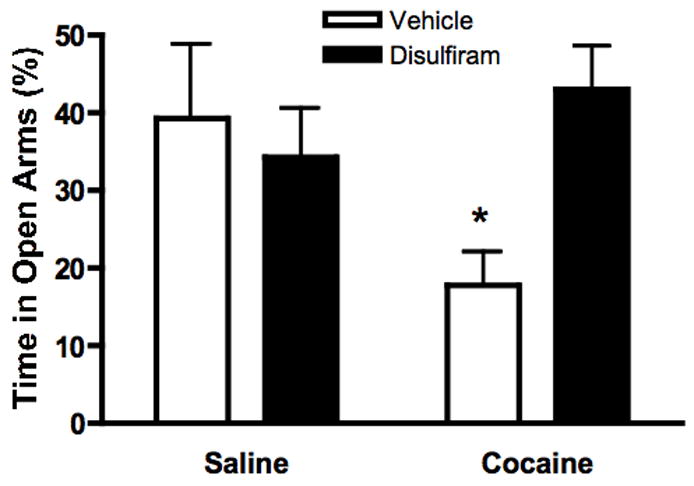
Disulfiram attenuates cocaine-induced anxiety in Dbh +/− mice. Dbh +/− mice were injected with disulfiram (3 × 200 mg/kg, i.p., two hours between each injection) or vehicle. Two hours following the last disulfiram treatment, mice were injected with saline or cocaine (10 mg/kg, i.p.), and tested in the EPM 20 minutes later. Shown is percent open arm time during the five minute EPM test. *p < 0.05 compared to Vehicle-Saline group (N=8 per group).
Blockade of β-adrenergic receptors attenuates cocaine-induced anxiety in control mice
The results of the preceding experiments indicated that NE is likely required for the anxiogenic effect of cocaine in the EPM. To determine which subtype of adrenergic receptor is critical for cocaine-induced anxiety, we pretreated Dbh +/− mice with the α1-AR antagonist prazosin, the α2-AR antagonist yohimbine, or the β-AR antagonist propranolol prior to administration of cocaine and EPM testing. We found that cocaine-induced anxiety was preserved following prazosin or yohimbine treatment, but abolished by propranolol (Fig. 4). One way ANOVA revealed a significant effect of antagonist treatment on percent open arm time (F[3,63]=3.485, p=0.0211), and Dunnett’s post-hoc tests indicated that the propranolol group differed significantly from the control group (p<0.01), whereas the prazosin and yohimbine groups did not. This suggests that NE signaling through β-adrenergic receptors is required for the cocaine-induced anxiety behavior of mice as measured by the EPM. To examine the possibility that the pretreatments alone can alter plus maze behavior, we treated Dbh +/− mice with either propranolol, prazosin, or yohimbine, at the same doses considered above, and tested plus maze behavior 20 minutes later. This data was then compared to behavior observed in saline-treated Dbh +/− mice, and one-way ANOVA revealed no significant effect of antagonist treatment on percent open time (F[3,31]=1.892, p=0.1539, n=7–10 per group).
Figure 4.
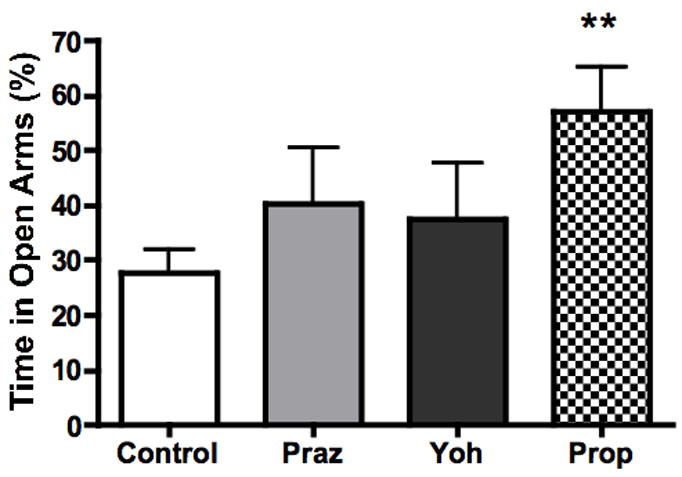
The β-adrenergic antagonist propranolol attenuates cocaine-induced anxiety. Dbh +/− mice were treated with vehicle, the α1-adrenergic antagonist prazosin, the α2-adrenergic antagonist yohimbine, or the β-adrenergic antagonist propranolol 10 minutes prior to cocaine injection (10 mg/kg, i.p.), and mice were tested in the EPM 20 minutes later. Shown is percent open arm time during the five minute EPM test. ** p < 0.01 compared to vehicle control (N=10–17 per group).
Blockade of β-adrenergic receptors attenuates cocaine-induced anxiety in C57BL6/J mice
To assess whether our results could be generalized to other wild-type mouse strains, we tested the effect of propranolol pretreatment on cocaine-induced anxiety behavior in pure C57BL6/J mice. Similar to our previous results, cocaine reduced percent open arm time, and this effect was blocked by propranolol. Propranolol had no effect on baseline performance. Two way ANOVA revealed a main effect of drug treatment (F[1,24]=5.694, p=0.0253). Bonferroni post-hoc tests indicated that only percent open arm time of the Vehicle-Cocaine group was significantly lower than the Vehicle-Saline group (p<0.05).
Discussion
Although cocaine-induced anxiety is widely recognized in both animals and humans, very little is known about the mechanism underlying these effects. The main finding of our current experiments is that a complete lack of NE, such as occurs in Dbh −/− mice, abolishes cocaine-induced anxiety. This phenotype was mimicked by either inhibition of DBH activity or β-AR signaling in control animals. In these experiments, we used percent open time as our dependent variable to account for any effects of genotype or drug treatment on exploratory activity. Our findings therefore imply a specific role for NE in cocaine-induced anxiety.
We used selective adrenergic antagonists in control mice to determine which subtype of adrenergic receptor is critical for cocaine-induced anxiety. We found that propranolol, but not prazosin or yohimbine, recapitulated the Dbh −/− phenotype and prevented the cocaine-induced decrease in open arm time. These results indicate that activation of β-adrenergic receptors is required for the anxiogenic effects of cocaine in this test. The behavioral effect of β-adrenergic blockade was replicated in wild-type C57BL6/J mice, demonstrating that the results observed are not specific to our Dbh animals.
The specific location of the β-adrenergic receptors necessary for mediating cocaine-induced anxiety is not known. While the striatum and prefrontal cortex appear to be important for mediating the locomotor-activating and rewarding effects of cocaine, it seems likely that cocaine-induced anxiety is mediated elsewhere. The amygdala and bed nucleus of the stria terminalis are appealing candidates because of the dense noradrenergic innervation they receive and their involvement in general and drug-specific stress responses (23–27).
One of the initial motives driving these experiments was to determine if the hypersensitivity to cocaine-induced place aversion observed in Dbh −/− mice might be accounted for by increases in cocaine-induced anxiety. Our results, however, reveal a dissociation between the anxiogenic effects of cocaine in the EPM and the aversive effects of cocaine in the place conditioning test. While Dbh −/− mice find a 20 mg/kg dose of cocaine strongly aversive in a place conditioning paradigm (9), they were completely insensitive to the anxiogenic properties of the same dose of cocaine in the EPM in this study. We conclude that some unpleasant effect of cocaine, aside from increased anxiety, must account for the hypersensitivity to the aversive effects of cocaine seen in Dbh −/−mice. Human cocaine addicts with genetically low DBH activity experience more severe cocaine-induced paranoia than individuals with high DBH activity, while anxiety measures are unaffected by DBH genotype (28,29). It is unclear whether rodents can experience anything resembling human paranoia, but an analogous negative state may perhaps contribute to cocaine aversion in Dbh −/− mice. Dbh −/− mice are also profoundly hypersensitive to the locomotor activating effects of cocaine, which appears to be mediated by the amplified dopamine receptor signaling that develops during chronic NE deficiency, rather than a specific lack of NE at the time of cocaine administration (9). In contrast, the insensitivity of Dbh −/− mice to the anxiogenic effects of cocaine is due to an “acute” lack of NE and is unrelated to excessive dopamine signaling.
Another dissociation appears to apply to adrenergic receptor systems involved in these drug responses. While the α1-adrenergic receptor has been identified as the key modulator of the locomotor activating and rewarding effects of cocaine (8,30), the α1-adrenergic receptor antagonist prazosin was without effect in our EPM experiments; only the β-adrenergic antagonist propranolol led to a reduction in cocaine-induced anxiety-like behavior in control mice. These results demonstrate the involvement of distinct receptor systems in the effects of NE on drug-induced hyperactivity/reward and anxiety behaviors in mice.
We found that acute restoration of NE to Dbh −/− mice using DOPS also restored cocaine-induced anxiety in the EPM. However, DOPS also tended to decrease percent open arm time in saline-treated Dbh −/− mice, complicating the interpretation of these results. An important consideration that might explain this result is that Dbh −/− mice lack NE from birth and have a compensatory increase in brain β-adrenergic receptors (31). Thus, it is possible that the acute introduction of NE produces a surge in β-adrenergic signaling in Dbh −/− mice analogous to that produced by cocaine in normal animals, and increases anxiety-like behavior. Although this effect makes it difficult to draw conclusions about cocaine-induced anxiety in this particular experiment, our results with disulfiram and propranolol in control mice support the conclusion that the anxiogenic effects of cocaine require NE and β-adrenergic signaling.
Our results are pertinent to the clinical treatment of drug dependence because the DBH inhibitor disulfiram is capable of decreasing cocaine intake in human addicts (32). We found that disulfiram abolishes the effects of cocaine in the EPM, which is counterintuitive because one would expect a treatment that reduces one of the aversive effects of cocaine to encourage drug use, rather than attenuate it. Notably, however, cocaine withdrawal also produces anxiety, which often precipitates relapse. While our study does not specifically address chronic cocaine use and subsequent withdrawal and relapse effects, the β-AR also appears to play a vital role in the expression of cocaine withdrawal-induced anxiety (33–36) and stress-induced reinstatement (25). Combined with the data presented here, these results suggest that the acute anxiogenic effects of cocaine and the anxiety associated with cocaine withdrawal depend on a similar β-adrenergic receptor mediated signaling pathway. Thus, we hypothesize that the ability of disulfiram to reduce cocaine intake in addicts is due, in part, to a reduction in NE synthesis and subsequent attenuation of withdrawal-associated anxiety and stress-induced relapse. Therefore, β-adrenergic receptor antagonists may also be useful as cocaine addiction pharmacotherapy by interfering with these anxiety responses.
Figure 5.
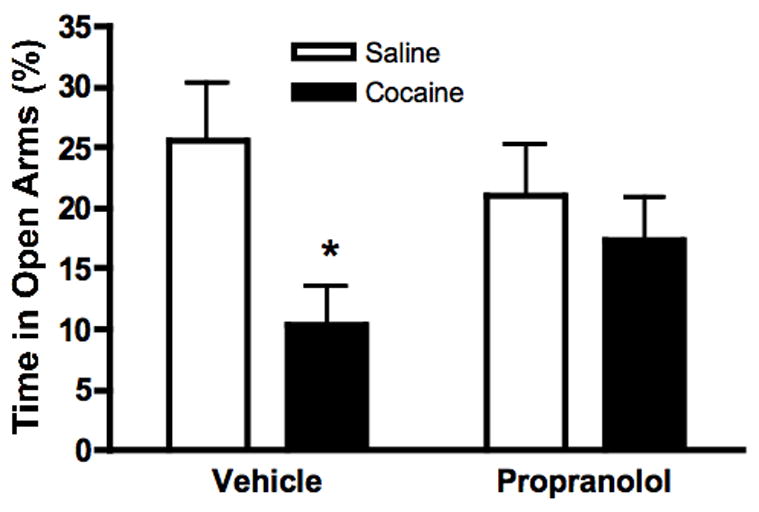
The β-adrenergic antagonist propranolol attenuates cocaine-induced anxiety in wild type C57BL6/J mice. Wild-type C57BL6/J mice were treated with either propranolol or saline 10 minutes prior to cocaine injection, and mice were tested on the EPM 20 mniutes later. Shown is percent open time during the five minute EPM test. *p<0.05 compared to saline control (N=7 per group).
Acknowledgments
We thank Dainippon-Sumitomo Pharma Co. Ltd. (Osaka, Japan) for their generous donation of DOPS, and Cheryl Strauss for assistance in text editing. We also thank Dr. Larry Young (Emory University) for use of the elevated plus maze apparatus. This work was funded by NIH/NIDA (DA017963, DA019849), and J.R.S received support from the Emory University Training Program in Drug Abuse (DA015040) and Predoctoral NRSA (DA019746).
Footnotes
Financial Disclosure
We have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ettenberg A, Geist TD. Animal model for investigating the anxiogenic effects of self-administered cocaine. Psychopharmacology. 1991;103:455–461. doi: 10.1007/BF02244244. [DOI] [PubMed] [Google Scholar]
- 2.Rogiero R, Takahashi RN. Anxiogenic properties of cocaine in the rat evaluated with the elevated plus maze. Pharmacology, Biochemistry, and Behavior. 1992;43:631–633. doi: 10.1016/0091-3057(92)90203-r. [DOI] [PubMed] [Google Scholar]
- 3.Yang X-M, Gorman AL, Dunn AJ, Goeders NE. Anxiogenic Effects of Acute and Chronic Cocaine Administration: Neurochemical and Behavioral Studies. Pharmacology Biochemistry and Behavior. 1992;41:643–650. doi: 10.1016/0091-3057(92)90386-t. [DOI] [PubMed] [Google Scholar]
- 4.Costall B, Kelly E, Naylor RJ, Onaivi ES. The actions of nicotine and cocaine in a mouse model of anxiety. Pharmacology, Biochemistry, and Behavior. 1988;33:197–203. doi: 10.1016/0091-3057(89)90450-4. [DOI] [PubMed] [Google Scholar]
- 5.Blanchard DC, Blanchard RJ. Cocaine potentiates defensive behaviors related to fear and anxiety. Neuroscience and Biobehavioral Reviews. 1999;23:981–991. doi: 10.1016/s0149-7634(99)00031-7. [DOI] [PubMed] [Google Scholar]
- 6.David V, Gold LH, Koob GF, Cazala P. Anxiogenic-like effects limit rewarding effects of cocaine in BALB/cByJ Mice. Neuropsychopharmacology. 2001;24(3):300–318. doi: 10.1016/S0893-133X(00)00205-0. [DOI] [PubMed] [Google Scholar]
- 7.Paine TA, Jackman SL, Olmstead MC. Cocaine-induced anxiety: alleviation by diazepam, but not buspirone, dimenhydrinate, or diphenhydramine. Behavioural Pharmacology. 2002;13:511–523. doi: 10.1097/00008877-200211000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Drouin C, Darracq L, Trovero F, Blanc G, Glowinski J, Cotecchia S, Tassin J-P. α1b-adrenergic receptors control locomotor and rewarding effects of psychostimulants and opiates. Journal of Neuroscience. 2002;22:2873–2884. doi: 10.1523/JNEUROSCI.22-07-02873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schank JR, Puglisi-Allegra S, Ventura R, Alcaro A, Cole CD, Liles LC, Seeman P, Weinshenker D. Dopamine β-hydroxylase knockout mice have alterations in dopamine signaling and are hypersensitive to cocaine-induced locomotion, reward, and aversion. Neuropsychopharmacology. 2006;31:2221–2230. doi: 10.1038/sj.npp.1301000. [DOI] [PubMed] [Google Scholar]
- 10.Ventura R, Cabib S, Alcaro A, Orsini C, Puglisi-Allegra S. Norepinephrine in the prefrontal cortex is critical for amphetamine-induced reward and mesoaccumbens dopamine release. Journal of Neuroscience. 2003;23:1879–1885. doi: 10.1523/JNEUROSCI.23-05-01879.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinshenker D, Miller NS, Blizinsky K, Laughlin ML, Palmiter RD. Mice with chronic norepinephrine deficiency resemble amphetamine-sensitized animals. PNAS. 2002;99(21):13873–13877. doi: 10.1073/pnas.212519999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorman AL, Dunn AJ. β-Adrenergic receptors are involved in stress-related behavioral changes. Pharmacology Biochemistry and Behavior. 1993;45:1–7. doi: 10.1016/0091-3057(93)90078-8. [DOI] [PubMed] [Google Scholar]
- 13.Stanford SC. Central noradrenergic neurons and stress. Pharmacology and Therapeutics. 1995;68:297–342. doi: 10.1016/0163-7258(95)02010-1. [DOI] [PubMed] [Google Scholar]
- 14.Thomas SA, Matsumoto AM, Palmiter RD. Noradrenaline is essential for mouse fetal development. Nature. 1995;374:643–646. doi: 10.1038/374643a0. [DOI] [PubMed] [Google Scholar]
- 15.Thomas SA, Marck BT, Palmiter RD, Matsumoto AM. Restoration of Norepinephrine and Reversal of Phenotypes in Mice Lacking Dopamine β-Hydroxylase. Journal of Neurochemistry. 1998;70:2468–2476. doi: 10.1046/j.1471-4159.1998.70062468.x. [DOI] [PubMed] [Google Scholar]
- 16.Bourdelat-Parks BN, Anderson GM, Donaldson ZR, Weiss JM, Bonsall RW, Emery MS, Liles LC, Weinshenker D. Effects of dopamine beta-hydroxylase genotype and disulfiram inhibition on catecholamine homeostasis in mice. Psychopharmacology. 2005;183(1):72–80. doi: 10.1007/s00213-005-0139-8. [DOI] [PubMed] [Google Scholar]
- 17.Pellow S, Chopin P, File SE, Briley M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. Journal of Neuroscience Methods. 1985;14:149–167. doi: 10.1016/0165-0270(85)90031-7. [DOI] [PubMed] [Google Scholar]
- 18.Johnston AL, File SE. Yohimbine’s anxiogenic action: evidence for noradrenergic and dopaminergic sites. Pharmacology, Biochemistry, and Behavior. 1988;32:151–156. doi: 10.1016/0091-3057(89)90225-6. [DOI] [PubMed] [Google Scholar]
- 19.Murchinson CF, Zhang XY, Zhang WP, Ouyang M, Lee A, Thomas SA. A distinct role for norepinephrine in memory retrieval. Cell. 2004;117(1):131–143. doi: 10.1016/s0092-8674(04)00259-4. [DOI] [PubMed] [Google Scholar]
- 20.Maj J, Przegalinski E, Wielosz M. Disulfiram and the drug-induced effects on motility. Journal of Pharmacy and Pharmacology. 1968;20:247–248. doi: 10.1111/j.2042-7158.1968.tb09735.x. [DOI] [PubMed] [Google Scholar]
- 21.Musacchio JM, Goldstein M, Anagnoste B, Poch G, Kopin IJ. Inhibition of dopamine-beta-hydroxylase by disulfiram in vivo. Journal of Pharmacology and Experimental Therapeutics. 1966;152(1):56–61. [PubMed] [Google Scholar]
- 22.Marino MD, Bourdelat-Parks BN, Liles LC, Weinshenker D. Genetic reduction of noradrenergic function alters social memory and reduces aggression in mice. Behavioral Brain Research. 2005;161(2):197–203. doi: 10.1016/j.bbr.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 23.Erb S, Hitchcott PK, Rajabi H, Mueller D, Shaham Y, Stewart J. Alpha-2 Adrenergic Receptor Agonists Block Stress-Induced Reinstatement of Cocaine Seeking. Neuropsychopharmacology. 2000;23(2):138–150. doi: 10.1016/S0893-133X(99)00158-X. [DOI] [PubMed] [Google Scholar]
- 24.Shaham Y, Highfield D, Delfs J, Leung S, Stewart J. Clonidine blocks stress-induced reinstatement of heroin seeking in rats: an effect independent of locus coeruleus noradrenergic neurons. European Journal of Neuroscience. 2000;12(1):292–302. doi: 10.1046/j.1460-9568.2000.00899.x. [DOI] [PubMed] [Google Scholar]
- 25.Leri F, Flores J, Rodaros D, Stewart J. Blockade of Stress-Induced But Not Cocaine-Induced Reinstatement by Infusion of Noradrenergic Antagonists into the Bed Nucleus of the Stria Terminalis or the Central Nucleus of the Amygdala. Journal of Neuroscience. 2002;22(13):5713–5718. doi: 10.1523/JNEUROSCI.22-13-05713.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aston-Jones G, Harris GC. Brain substrates for increased drug seeking during protracted withdrawal. Neuropharmacology. 2004;47(Suppl 1):167–179. doi: 10.1016/j.neuropharm.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 27.Morilak DA, Barrera G, Echevarria DJ, Garcia AS, Hernandez A, Ma S, Petre CO. Role of brain norepinephrine in the behavioral response to stress. Progress in Neuropsychopharmacology and Biological Psychiatry. 2005;29(8):1214–1224. doi: 10.1016/j.pnpbp.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 28.Cubells JF, Kranzler HR, McCance-Katz E, Anderson GM, Malison RT, Price LH, Gelernter J. A haplotype at the DBH locus, associated with low plasma dopamine β-hydroxylase activity, also associates with cocaine-induced paranoia. Molecular Psychiatry. 2000;5:56–63. doi: 10.1038/sj.mp.4000657. [DOI] [PubMed] [Google Scholar]
- 29.Kalayasiri R, Sughondhabirom A, Gueorguieva R, Coric V, Lynch WJ, Lappalainen J, Gelernter J, Cubells JF, Mailson RT. Dopamine beta-hydroxylase gene (DbetaH)-1021C-->T influences self-reported paranoia during cocaine self-administration. Biological Psychiatry. 2006 Dec 5; doi: 10.1016/j.biopsych.2006.08.012. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 30.Darracq L, Blanc G, Glowinski J, Tassin JP. Importance of the noradrenaline-dopamine coupling in the locomotor activating effects of D-amphetamine. Journal of Neuroscience. 1998;18(7):2729–2739. doi: 10.1523/JNEUROSCI.18-07-02729.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanders JD, Szot P, Weinshenker D, Happe HK, Bylund DB, Murrin LC. Analysis of brain adrenergic receptors in dopamine β-hydroxylase knockout mice. Brain Research. 2006;1109:45–53. doi: 10.1016/j.brainres.2006.06.033. [DOI] [PubMed] [Google Scholar]
- 32.Carroll KM, Fenton LR, Ball SA, Nich C, Frankforter TL, Shi J, Rounsaville BJ. Efficacy of Disulfiram and Cognitive Behavior Therapy in Cocaine-Dependent Outpatients. Archives of General Psychiatry. 2004;61:264–272. doi: 10.1001/archpsyc.61.3.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kampman KM, Volpicelli JR, Mulvaney F, Alterman AI, Cornish J, Gariti P, Cnaan A, Poole S, Muller E, Acosta T, Luce D, O’Brien C. Effectiveness of propranolol for cocaine dependence treatment may depend on cocaine withdrawal symptom severity. Drug and Alcohol Dependence. 2001;63:89–78. doi: 10.1016/s0376-8716(00)00193-9. [DOI] [PubMed] [Google Scholar]
- 34.McDougle CJ, Black JE, Malison RT, Zimmermann RC, Kosten TR, Heninger GR, Price LH. Noradrenergic dysregulation during discontinuation of cocaine use in addicts. Archives of General Psychiatry. 1994;51(9):713–719. doi: 10.1001/archpsyc.1994.03950090045007. [DOI] [PubMed] [Google Scholar]
- 35.Harris GC, Aston-Jones G. Beta-adrenergic antagonists attenuate withdrawal anxiety in cocaine- and morphine-dependent rats. Psychopharmacology. 1993;113(1):131–136. doi: 10.1007/BF02244345. [DOI] [PubMed] [Google Scholar]
- 36.Rudoy CA, Van Bockstaele EJ. Betaxolol, a selective β-adrenergic receptor antagonist, diminishes anxiety-like behavior during early withdrawal from chronic cocaine administration in rats. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2007;31:1119–1129. doi: 10.1016/j.pnpbp.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]