

Abstract
Glycine N-methyltransferase (GNMT) is the main enzyme responsible for catabolism of excess hepatic S-adenosylmethionine (SAMe). GNMT is absent in hepatocellular carcinoma (HCC), messenger RNA (mRNA) levels are significantly lower in livers of patients at risk of developing HCC, and GNMT has been proposed to be a tumor-susceptibility gene for liver cancer. The identification of several children with liver disease as having mutations of the GNMT gene further suggests that this enzyme plays an important role in liver function. In the current study we studied development of liver pathologies including HCC in GNMT-knockout (GNMT-KO) mice. GNMT-KO mice have elevated serum aminotransferase, methionine, and SAMe levels and develop liver steatosis, fibrosis, and HCC. We found that activation of the Ras and Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathways was increased in liver tumors from GNMT-KO mice coincidently with the suppression of the Ras inhibitors Ras-association domain family/tumor suppressor (RASSF) 1 and 4 and the JAK/STAT inhibitors suppressor of cytokine signaling (SOCS) 1-3 and cytokine-inducible SH2-protein. Finally, we found that methylation of RASSF1 and SOCS2 promoters and the binding of trimethylated lysine 27 in histone 3 to these 2 genes was increased in HCC from GNMT-KO mice.
Conclusion
These data demonstrate that loss of GNMT induces aberrant methylation of DNA and histones, resulting in epigenetic modulation of critical carcinogenic pathways in mice.
The first steps in mammalian methionine metabolism are conversion to S-adenosylmethionine (SAMe) and transfer of the methyl group of SAMe to a large variety of substrates (including DNA, RNA, histones, and small molecules such as glycine, guanidinoacetate, and phosphatidylethanolamine) with the formation of S-adenosylhomocysteine (SAH), an inhibitor of many SAMe-dependent methyltransferases.1 Although there are a large number of SAMe-dependent methyltransferases,2 methylation of glycine by glycine N-methyltransferase (GNMT) to form sarcosine (N-methylglycine) is one of the reactions that contribute most to total transmethylation flux.3 The importance of GNMT is to remove excess SAMe and maintain a constant hepatic SAMe/SAH ratio to avoid aberrant methylation.2 Consistent with this function, the activation of GNMT in rats by the administration of retinoic acid causes a reduction in plasma methionine and homocysteine levels, as well as in liver DNA methylation.4,5 In GNMT-knockout (GNMT-KO) mice, liver SAMe content is elevated 35-fold, and the SAMe/SAH ratio increases about 100-fold,6 and individuals with GNMT mutations, which leads to inactive forms of the enzyme, have elevated plasma levels of methionine and SAMe but a normal concentration of homocysteine.7,8
GNMT is expressed in the liver, pancreas, and prostate9 and is absent in hepatocellular carcinoma (HCC)10 and down-regulated in the livers of patients at risk of developing HCC, such as in those with hepatitis C virus–and alcohol-induced cirrhosis.11 Moreover, a loss of heterozygosity of GNMT has been reported in about 40% of HCC patients, and GNMT has been proposed to be a tumor-susceptibility gene for liver cancer.12,13 Identification of several children with mutations of GNMT as having mild to moderate liver disease with elevated serum aminotransferases7,8 further suggests that a defect in this enzyme leads to liver disease.
To define the role of GNMT in the liver, we examined hepatic function in GNMT-KO mice. The GNMT-deficient mice recapitulated the situation observed in individuals with mutations in GNMT7,8 and showed elevated serum aminotransferases, methionine, and SAMe levels. Moreover, the GNMT-KO mice developed steatosis, fibrosis, and HCC. Finally, we analyzed the activation of the Ras and Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathways in GNMT-KO mice, as well as the methylation of DNA and H3K27, which may result in epigenetic modulation of critical carcinogenic pathways.
Materials and Methods
Animal Experiments
The generation of GNMT-KO mice has been described.6 All animal experimentation was conducted in accordance with the Spanish Guide for the Care and Use of Laboratory Animals, and protocols were approved by the CIC bioGUNE Ethical Review Committee. Three-month-old and 8-month-old male homozygous GNMT-KO and their wild-type (WT) littermates were killed for histological examination of their livers. In addition, liver specimens were snap-frozen for subsequent analysis. At the time of death, serum samples were collected for determination of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels or deproteinized with acetonitrile for methionine and SAMe analysis.
Measurement of Methionine and SAMe
Serum levels of methionine and SAMe were determined by liquid chromatography/mass spectrometry (LC/MS) using a Waters Acquity ultraperformance liquid chromatography (UPLC) system equipped with a column oven, and a Waters Acquity UPLC BEH C18 column (2.1 × 100 mm, 1.7 μm). A 3-μL aliquot of sample was injected into the UPLC column, which was maintained at 50°C and eluted at 600 μL/min. The mobile phase consisted of water with 0.05% formic acid (solvent A) and acetonitrile with 0.05% formic acid (solvent B). Separation was carried out starting with 1% B for 1 minute, followed by a linear gradient from 1% to 95% B for 10 minutes and staying at 95% B for 6 minutes. The UPLC system was coupled to a Waters Micromass LCT Premier Mass Spectrometer equipped with a Lockspray ionization source operating in electrospray positive ion mode (W mode, resolution 10,000 full width at half maximum). Full-scan time-of-flight MS spectra were acquired between m/z (mass-to-charge ratio) 80 and 1,000 using the dynamic range enhancement mode, with a total scan time of 0.4 seconds. Leucine enkephalin was used as a reference compound for accurate mass measurements. The reference was infused into the Lockspray reference channel, and the masses of the analyte channel were automatically mass-corrected by the software. A set of samples, prepared from all standard compounds, was acquired over the concentration range of 10 pg/μL to 50 ng/μL in order to test the linearity of the system. After analyzing all concentration levels, calibration curves were generatedμL and chromatogram traces of the protonated species of the standards were extracted with a mass window of 0.05 Da. Serum levels of methionine and SAMe were automatically calculated from the calibration curves.
Histology and Immunohistochemistry
Sections from formalin-fixed liver tissue were stained with hematoxylin and eosin or with Sirius red for collagen visualization. The proliferation marker Ki67 was evaluated by immunostaining using mouse monoclonal anti-Ki67 primary antibody.
Quantitative Real-Time PCR
Total liver RNA was isolated by the guanidinium thiocyanate method. RNA concentration was determined spectrophotometrically before use, and integrity was checked by electrophoresis with subsequent ethidium bromide staining. The RNA was purified with an RNeasy Mini kit (Qiagen, Chatsworth, CA) including DNase treatment. Two micrograms of total RNA were then retrotranscribed with the M-MLV retrotranscriptase (Promega, Madison, WI) in the presence of an oligo dT. We performed quantitative real-time polymerase chain reaction (PCR) in a Bio-Rad iCycler thermalcycler (Bio-Rad Laboratories, Hercules, CA). All samples were run in triplicate. Five microliters of a 1:20 dilution of cDNA was used in each PCR reaction in a total reaction volume of 30 μL using iQ SYBR Green Super Mix (Bio-Rad). Primers were designed with Primer Express Software (Applied Biosystems, Foster City, CA) to determine the expression of Ras-association domain family/tumor suppressor (RASSF)–1, RASSF4, cytokine-inducible SH2-protein (CIS), SOCS1, SOCS2, SOCS3, ADFP, UCP2, CYP4A10, CYP4A14, TIMP-1, and iNOS and synthesized by Invitrogen. After checking the specificity of the PCR products with the melting curve, Ct values were extrapolated to a standard curve performed simultaneously with the samplesμL and data were then normalized to GAPDH expression. The complete list of primer sequences used and the PCR conditions are available on request.
Western Blotting
Samples were separated by SDS-PAGE and analyzed by immunoblotting using commercial antibodies. Blots were developed with secondary antirabbit or antimouse antibodies conjugated to horseradish peroxidase (Invitrogen, Carlsbad, CA) and the luminal-chemiluminescence reagent (ECL; Amersham Biosciences, Upsala, Sweden). The processed blots were exposed to X-ray film, and the autoradiograms were analyzed.
Ras Activity
Liver tissue samples were homogenized in lysis buffer [125 mM HEPES (pH 7.5), 750 mM NaCl, 5% Igepal CA-630, 50 mM MgCl2, 5 mM EDTA, and 10% glycerol] containing Complete Protease Inhibitor Cocktail (Roche Molecular Biochemicals) and sonicated. Protein concentrations were determined with a Bio-Rad Protein Assay Kit using bovine serum albumin as a standard. Aliquots of 800 μg of tissue lysates were immunoprecipitated with 5 μg of mouse monoclonal anti–pan Ras antibody (sc-32; Santa Cruz Biotechnology, Santa Cruz, CA). Immunoprecipitated proteins were denatured by boiling in Tris-Glycine SDS Sample Buffer (Novex-Invitrogen, Carlsbad, CA), separated by SDS-PAGE, and then transferred onto nitrocellulose membranes (Novex-Invitrogen) by electroblotting. The membranes were blocked in 5% nonfat dry milk in Tris-buffered saline containing 0.1% Tween 20 for 1 hour and probed with the rabbit polyclonal anti-RAF-1 antibody (sc-133, Santa Cruz Biotechnology) in a 1:2,000 dilution. Immunoprecipitated proteins were revealed via enhanced chemiluminescence (Amersham), and the bands were quantified in arbitrary units by ImageMaster Total Lab V1.11 software and normalized to actin levels (as detected with the rabbit polyclonal antiactin antibody sc-10731; Santa Cruz Biotechnology).
Global DNA Methylation and DNA Methyltransferase Activity
5-Methylcytosine (5mC) genomic content was determined by high-performance capillary electrophoresis, as described.14 Briefly, genomic DNA samples were boiled, treated with nuclease P1 (Sigma) for 16 hours at 37°C and with alkaline phosphatase (Sigma) for an additional 2 hours at 37°C. After hydrolysis, total cytosine and 5mC content were measured by capillary electrophoresis using a P/ACE MDQ system (Beckman-Coulter). All samples were analyzed in triplicate. Relative 5mC content was expressed as a percentage of total cytosine content (methylated and nonmethylated). DNA methyltransferase activity was measured using an EpiQuick DNA methyltransferase Activity kit.
Sequence-Specific DNA Methylation
The CpG DNA methylation status of a subtelomeric DNA region at chromosome 1 and of the promoters of RASSF1 and SOCS2 was established by PCR analysis after bisulfite modification. Bisulfite genomic sequencing of multiple clones was then carried out as described elsewhere.15 The primers used were: 5′-AAAAGATGGTGAGTGTAGGTGT-3′ (sense) and 5′-TCACAATATCCTCCTCTTCTTC-3′ (antisense) for chromosome 1; 5′-CGAACGTGGTGAGCGCGG-3′(sense) and 5′-CGCCCCTCGACGTCTTCACCCCCTCAC-3′ (antisense) for RASSF1; and 5′-TGTCTCCCTCCTCACCCCTCTACTCCGG-3′ (sense) and 5′-ACGCGAGGTCCACTGGCCCCAGC-3′ (antisense) for SOCS2.
Promoter Methylation
MspI and HpaII restriction endonucleases were used as described with modifications.16 MspI and HpaII can distinguish between unmethylated and methylated cytosine in the nucleotide sequence 5′-CCGG. MspI is insensitive to the methylation status, whereas HpaII will digest only if the internal cytosine is unmethylated.26 DNA samples (30 μg each) were first digested with StuI and BbvI for RASSF1 or BspmII and TthlllI for SOCS2 at 37°C for 4 hours. This was followed by overnight digestion with HpaII or MspI at 37°C. The digested DNA was separated on 1% agarose, transferred onto nylon filters by 20× standard sodium citrate (SSC), and cross-linked in an ultraviolet cross-linker (FS-UVXL-1000, Fisher Biotech). The filters were prehybridized at 42°C in Rapid-Hybridization Buffer (Ambion, Austin, TX) for 2 hours and hybridized with the [32P]dCTP-labeled RASSF1 probe (corresponding to −313 to −780 base pairs upstream of the mouse RASSF1 translational start site, GenBank accession number NM_019713) or the SOCS2 probe (corresponding to −205 to −511 base pairs upstream of the mouse SOCS2 translational start site, GenBank accession number NM_007706) for 8 hours at 42°C in the same buffer. The blots were washed at 42°C for 20 minutes in 2× SSC containing 0.1% SDS twice and at 42°C for 30 minutes in 0.1×SSC containing 0.1% SDS. Autoradiography was performed by exposure to Kodak BioMax MR film at −80°C.
Analysis of Trimethylated Lysine 27 in Histone 3
Chromatin immunoprecipitation assay was carried out as described15 using antibodies against trimethylated lysine 27 in histone 3 (H3K27me3; Abcam). Chromatin was sheared to an average length of 0.25 ± 1 kb, and PCR amplification was performed in 96-well optical plates in a volume of 20 μL. We used 15 ng of immunoprecipitated DNA, 5 pmol of each primer, and 10 μL of 2× SYBRGreen PCR master Mix (Applied Biosystems). All measurements were carried out in triplicate, and amounts were calculated extrapolating from a standard curve. The primers and conditions for each promoter are available on request.
Statistical Analysis
The Student t test was used to evaluate statistical significance. P values < 0.05 were considered statistically significant.
Results
Deletion of GNMT Gene Induces Steatosis, Fibrosis, and Hepatocellular Carcinoma
To define the metabolic role of GNMT in the liver, we examined hepatic function in GNMT-KO and WT male mice. The GNMT-deficient mice recapitulated the situation observed in children with mutations in GNMT8,9 and showed elevated serum aminotransferases, methionine and SAMe at both 3 and 8 months of age (Table 1). Histological examination of the livers of 3-month-old mutant mice showed steatosis and fibrosis, which were more prominent in the livers of 8-month-old mice (Fig. 1A). No significant signs of inflammation were observed at either 3 or 8 months of age. At 8 months, all GNMT-KO mice (n = 10) also developed multifocal HCC, as determined by staining with hematoxylin and eosin and with the cellular proliferation marker Ki67 (Fig. 1). The number of liver tumors varied from 2 to 9, with an average of 4 tumors per mouse. No evidence of focal hyperplasic lesions was observed in GNMT-KO livers at 3 or 5 months of age. Histological examination of prostate, pancreas, and kidney did not reveal signs of tumor formation in 8-month-old GNMT-KO mice.
Table 1.
Serum Methionine (Met), S-Adenosylmethionine (SAMe), and Aminotransferases (ALT and AST) in 3-Month-Old and 8-Month-Old Wild-Type (WT) and GNMT-Deficient (GNMT-KO) Mice
| WT
(3 months) |
GNMT-KO
(3 months) |
WT
(8 months) |
GNMT-KO
(8 months) |
|
|---|---|---|---|---|
| Met (μmol/L) | 24.9 ± 2.7 | 81.9 ± 23.3* | 51.6 ± 7.5 | 151 ± 55.0* |
| SAMe (μmol/L) | 0.5 ± 0.05 | 0.9 ± 0.2* | 0.6 ± 0.1 | 1.2 ± 0.4* |
| ALT (IU/L) | 100 ± 26.6 | 166 ± 45.3* | 105 ± 35.6 | 320 ± 86.2* |
| AST (IU/L) | 47.5 ± 13.3 | 99.3 ± 35.8* | 76.6 ± 19.4 | 347 ± 134* |
Data are the means ± SDs. of at least 5 animals per group.
P < 0.05 WT versus GNMT-KO.
Fig. 1. Deletion of GNMT leads to steatosis, fibrosis, and HCC.
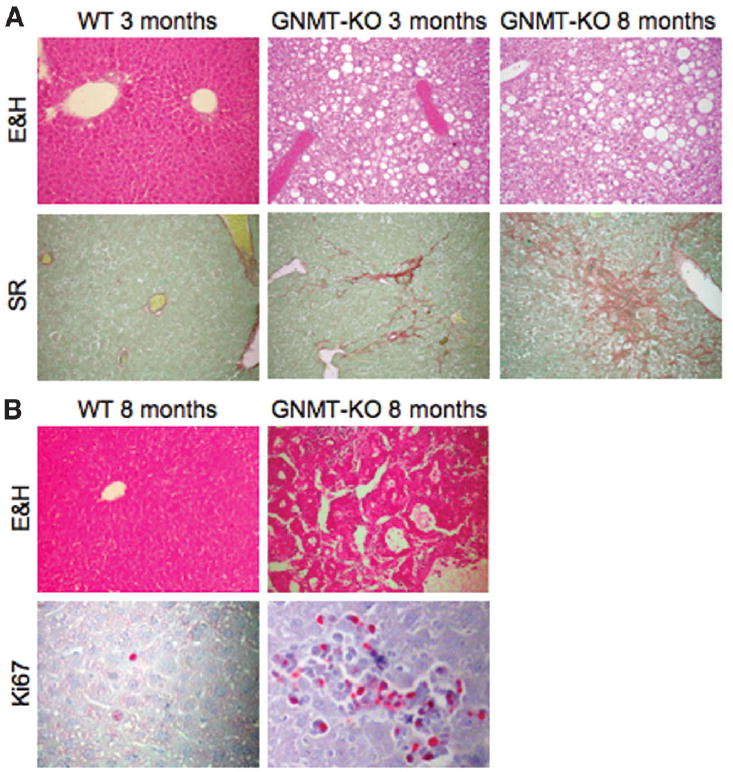
(A) At 3 months of age, macro and microvesicular steatosis (white droplets) could be seen through the hepatic lobule in GNMT-knockout (GNMT-KO) mice compared with wild-type (WT) animals. Collagen deposits (stained red) indicate moderate liver fibrosis. By 8 months of age, liver steatosis and fibrosis in mutant mice were more prominent. At least 10 animals per group were examined. (B) At 8 months of age, livers from GNMT-KO mice also had multifocal HCC. Liver cords up to 5 cells thick, lined by endothelial cells, could be seen, with occasional pseudogland formation. Staining with the nuclear proliferation marker Ki67 (red spots) indicates a higher proportion of cells in the cell cycle compared with WT cells. At least 10 animals per group were examined (E&H, eosin and hematoxylin staining; SR, Sirius red staining). Original magnification ×40.
Activation of Ras and JAK/STAT Pathways in HCC from GNMT-Deficient Mice
HCC in humans is characterized by the persistent activation of the Ras/mitogen-activated/extracellular-regulated kinase (MEK)/extracellular signal-regulated kinase (ERK) and JAK/STAT signaling pathways via suppression of Ras and JAK/STAT inhibitors such as RASSF1, CIS, and SOCS1, SOCS2, and SOCS3.17-21 Accordingly, we observed that the expression of Ras inhibitors RASSF1 and RASSF4 and of JAK/STAT inhibitors SOCS1, SOCS2, SOCS3, and CIS was reduced in liver tumors from 8-month-old GNMT-KO male mice (Table 2). Concomitant with the loss of Ras inhibitors, Ras and downstream effectors of Ras involved in proliferation and survival, including pRaf, pMEK1/2, and pERK1/2, were activated in liver tumors from GNMT-deficient mice (Fig. 2A). Ras activity, assessed by immunoprecipitation with anti–pan Ras antibody and probed with anti-RAF-1 antibody, was also markedly increased in liver tumors from 8-month-old GNMT-mutant mice (Fig. 2B). Similarly, suppression of JAK/ STAT inhibitors in liver tumors from GNMT-KO mice was associated with the activation of the JAK/STAT signaling pathway, including proliferation proteins cyclin D1 and cyclin D2 and the antiapoptotic protein Bcl-xL (Fig. 2C). In these experiments we analyzed the liver containing tumors but not the surrounding liver. We chose to compare tumors to age-matched WT controls because the surrounding liver tissue may also be abnormal, as suggested by the finding that the expression of Ras and JAK/STAT inhibitors was already reduced in liver samples from 3-month-old GNMT-KO male mice (Table 2) and by the observation that Ras activity was also markedly increased in 3-month-old GNMT-KO mice liver (Fig. 2B).
Table 2.
Levels of Ras and JAK/STAT Inhibitors in GNMT-Knockout Mice
| Gene | Magnitude of change in GNMT-KO versus WT mice at 3 months | P value | Magnitude of change in GNMT-KO versus WT mice at 8 months | P value |
|---|---|---|---|---|
| SOCS 1 | −2.1 | 0.007 | −1.6 | 0.040 |
| SOCS 2 | −1.8 | 0.027 | −2.5 | 0.002 |
| SOCS 3 | −2.2 | 0.021 | −4.3 | 0.001 |
| CIS | −2.2 | 0.017 | −4.1 | 0.040 |
| RASSF 1 | −1.9 | 0.042 | −1.6 | 0.024 |
| RASSF 4 | −1.7 | 0.0003 | −2.4 | 0.010 |
Levels of JAK/STAT (SOCS1, SOCS2, SOCS3, CIS) and Ras (RASSF1 and RASSF4) inhibitors were determined by quantitative RT-PCR in the livers of wild-type (WT) mice at 3 and 8 months of age, in liver samples from 3-month-old GNMT-knockout (GNMT-KO) mice, and in liver tumors from 8-month-old knockout mice, normalized using GAPDH expression as a housekeeping gene, and the ratio (magnitude of change) of GNMT-KO versus WT calculated. Data represent means of at least 5 animals per subgroup assayed in triplicate.
Fig. 2. Activation of Ras and JAK/STAT pathways in GNMT-mutant mice liver.
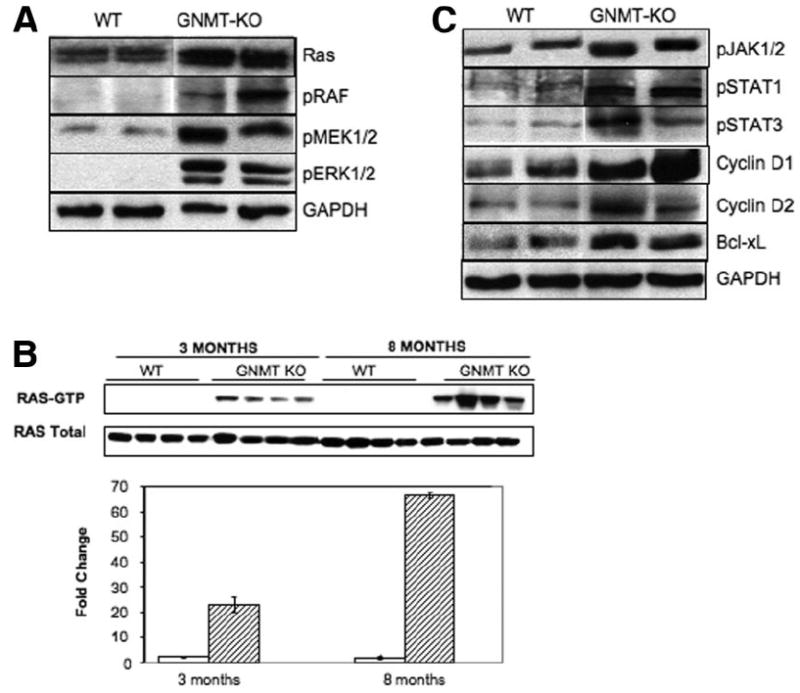
(A) Representative Western blot analysis of Ras downstream effectors (pRAF, pMEK1/2, and pERK1/2). Phosphorylation of RAF, MEK1/2, and ERK1/2 was markedly increased in liver tumors from GNMT-knockout (GNMT-KO) mice as compared with wild-type (WT) animals. One representative data set is shown out of 4 experiments performed. (B) Ras activity, assessed by immunoprecipitation with anti-pan Ras antibody and probed with anti-RAF-1 antibody, was markedly increased in livers from 3-month-old GNMT mice and in liver tumors from 8-month-old GNMT-mutant mice. Four animals from each subgroup were analyzed (WT mice, open bars; GNMT-KO mice, filled bars). (C) Representative Western blot analysis of JAK/STAT downstream effectors (pJAK1/2, pSTAT1, pSTAT3, cyclin D1 and D2, and Bcl-xL). Phosphorylation of JAK1/2, STAT1, and STAT 3 and protein levels of the JAK/STAT target genes, including the proliferation proteins cyclin D1 and cyclin D2 and the antiapoptotic protein Bcl-xL, were markedly increased in liver tumors from GNMT-KO mice. One representative data set is shown of 4 experiments performed. Unless otherwise indicated, animals were all 8 months old.
Increased Promoter Methylation and Binding to H3K27me3 of RASSF1 and SOCS2 in HCC from GNMT-KO Mice
Because the importance of GNMT is to remove excess SAMe and maintain a constant SAMe/SAH ratio to avoid aberrant methylation reactions,2 we hypothesized that the loss of GNMT may lead to abnormal DNA and histone methylation of specific and critical carcinogenic pathways. First, we assessed global DNA methylation and the methylation of a subtelomeric DNA region of chromosome 1 and found both to be hypermethylated in 8-month-old GNMT-KO male mice (Fig. 3A,B). Given that the inhibitors of the Ras and JAK/STAT signaling pathways are frequently inactivated by promoter methylation,17,19-21 we then examined the methylation of RASSF1 and SOCS2 promoters. Methylation-sensitive Southern blot analysis showed hypermethylation of these 2 promoters in liver tumors from 8-month-old GNMT-mutant mice (Fig. 3D,E). Because H3K27me3 is an epigenetic marker highly associated with genomic silencing,22 we also examined the trimethylation of H3K27 bound to RASSF1 and SOCS2 promoters. As shown in Fig. 3F,G, the level of H3K27me3 bound to RASSF1 and SOCS2 increased about 2-fold in liver tumors from GNMT-KO mice as compared with WT animals.
Fig. 3. Deletion of GNMT induces DNA and histone hypermethylation.
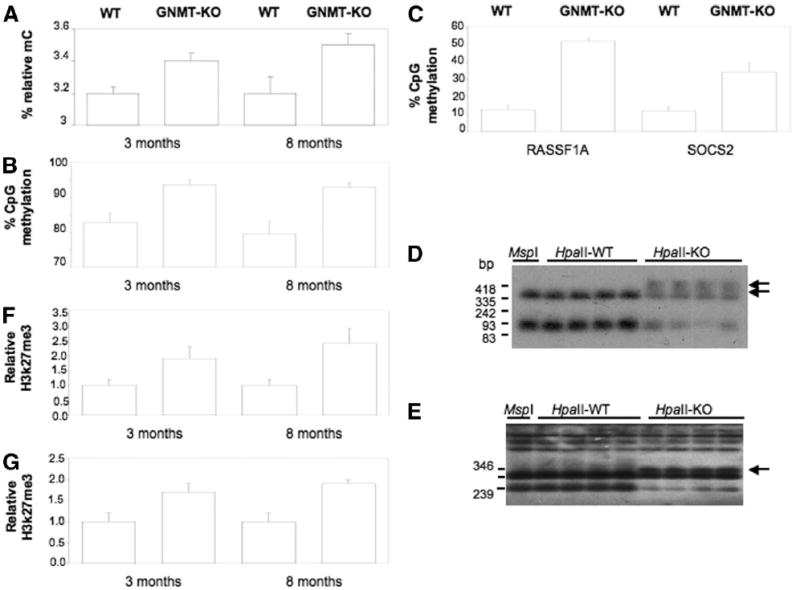
(A) Quantification of global DNA methylation. Total cytosine and 5-methylcytosine (5mC) content was determined in genomic DNA samples isolated from livers of wild-type (WT) mice at 3 and 8 months of age, in livers from 3-month-old GNMT-knockout (GNMT-KO) mice, and in liver tumors from 8-month-old knockout animals, and relative 5mC content, expressed as the percentage of total cytosine content (methylated and nonmethylated), was determined. Data represent means ± SDs from 5 animals per subgroup (*P < 0.05 versus WT). (B) Analysis of sequence-specific DNA methylation. CpG DNA methylation of a subtelomeric DNA region at chromosome 1 was determined by PCR analysis after bisulfite modification in samples isolated from livers of 3- and 8-month-old WT mice, in liver samples from 3-month-old GNMT-knockout mice, and in liver tumors from 8-month-old knockout animals. Data represent means ± SDs. from 5 animals per subgroup (*P < 0.05 versus WT mice). (c–e) Analysis of RASSF1 and SOCS2 promoter methylation. (C) To determine the CpG DNA methylation status of RASSF1 and SOCS2 promoters in liver samples from 3-month-old mice, we employed bisulfite genomic sequencing because the less sensitive technique, based on the use of MspI and HpaII restriction endonucleases, used at 8 month of age, did not reveal differences between GNMT-knockout and WT livers at 3 month of age. Data represent means ± SDs from 3 animals per subgroup (*P < 0.05 versus WT). (D, E) To analyze the methylation state of the promoters of RASSF1 and SOCS2 at 8 month of age, Southern blot analyses of genomic DNA digested with the methylation-sensitive isoschizomer restriction enzymes MspI and HpaII were performed as described in the Materials and Methods sections. Two MspI fragments were detected in Southern blots when hepatic DNA was hybridized with a RASSF1 probe (D). All DNA samples from WT mice liver digested with HpaII showed the same pattern of restriction fragments as that produced by MspI digestion, indicating that the CCGG sites were unmethylated. However, all DNA samples from GNMT-KO mice liver tumors digested with HpaII showed additional higher-molecular-weight bands (indicated by an arrow), showing that the RASSF1 promoter was hypermethylated. When hepatic DNA was hybridized with a SOCS2 probe (E), an additional band (indicated by arrows) was also detected when DNA from GNMT-KO liver tumors was digested with HpaII compared with when digested with MspI, whereas liver samples from WT mice showed the same pattern after digestion with either enzyme (HpaII-WT, DNA samples from WT liver mice digested with HpaII; HpaII-KO, DNA samples from GNMT-KO liver tumors digested with HpaII; 4 animals from each subgroup were analyzed). (F, G) Analysis of H3K27me3 bound to RASSF1 and SOCS2. The amount of H3K27me3 bound to RASSF1 (F) and SOCS2 (G) was determined by chromatin immunoprecipitation, as described in the Materials and Methods section in liver samples of 3- and 8-month-old WT mice, in liver samples of 3-month-old GNMT-knockout mice, and in liver tumors from 8-month-old knockout animals. Relative values of H3K27me3 are shown. Data represent means ± SDs from 5 animals per subgroup (*P < 0.05 versus WT).
Finally, we analyzed whether epigenetic pathways observed in the liver tumors were activated in 3-month-old mice before histological evidence of tumor development. As shown in Fig. 3, global DNA methylation, CpG DNA methylation of a subtelomeric DNA of chromosome 1, promoter methylation of RASSF1 and SOCS2 promoters, and trimethylation of H3K27 bound to RASSF1 and SOCS2 promoters were all significantly increased in liver samples from 3-month-old GNMT-deficient mice as compared to WT liver samples. To determine the CpG DNA methylation status of RASSF1 and SOCS2 promoters in liver samples from 3-month-old mice, we employed bisulfite genomic sequencing because the less sensitive technique used with livers from 8-month-old mice, based on the use of MspI and HpaII restriction endonucleases, did not reveal differences between GNMT-KO and WT livers from 3-month-old mice.
Liver DNA methyltransferase activity was similar in GNMT-deficient mice and WT animals at 3 month of age, whereas it was significantly increased (1.6 ± 0.2 fold, P < 0.05, n = 5) in the HCC nodules of 8-month-old GNMT-KO mice. These results suggest that epigenetic changes in GNMT-KO mice are most probably at least initially a result of the accumulation of SAMe and the increased SAMe/SAH ratio in the livers of these animals.
Discussion
GNMT is the most abundant methyltransferase in mammalian liver.9 GNMT catalyzes the conversion of glycine into sarcosine, which is then oxidized to regenerate glycine. The function of this futile cycle is to catabolize excess liver SAMe to maintain a constant SAMe/SAH ratio in order to avoid aberrant methylation reactions.1-3 Accordingly, the loss of GNMT in mice leads to the accumulation of hepatic SAMe and to a marked increase in the hepatic SAMe/SAH ratio without affecting SAH levels,6 and the activation of GNMT in rats by the administration of retinoic acid causes a reduction in global liver DNA methylation.4
In the present study, we showed that GNMT-deficient mice recapitulate the situation observed in individuals with mutations of the GNMT gene7,8 and have elevated serum aminotransferases, methionine, and SAMe. Moreover, in this study we showed that GNMT-deficient mice spontaneously develop steatosis, fibrosis, and HCC. These results support the concept that GNMT is a tumor-susceptibility gene for liver cancer12 and further suggest that reduced GNMT activity may be an early event in the development of HCC. Recently, the phenotype of a GNMT-KO mouse model that differs from ours was published.18 Consistent with the present results and with our previous observations,6 this other GNMT-KO mouse model has hypermethioninemia and elevated levels of both serum ALT and hepatic SAMe.18 However, these knockout mice did not develop steatosis or HCC, but about 60% of the animals had increased glycogen storage in the livers, as determined by staining with PAS.18 In the article by Liu et al.,18 no evidence about the molecular mechanism by which an increase in hepatic SAMe content induces glycogen storage is provided. In our GNMT-KO mouse model, hepatic PAS staining showed no differences between WT and knockout mice in glycogen staining (not shown). We believe that high SAMe levels can produce epigenetic changes that may differ depending on the genetic background and other factors such as nutritional factors, and it is possible this is why the phenotypes differ. Nevertheless, our finding of liver cancer development in GNMT-KO mice is consistent with the reported role of GNMT as a tumor suppressor in human hepatocellular carcinoma.
Previous studies have demonstrated that the Ras and JAK/STAT pathways are universally activated in human HCC and that the activation of these pathways may be essential for HCC development.17 We have observed that, as in human HCC, the Ras and JAK/STAT pathways are activated in liver tumors from GNMT-deficient mice and that, as also found in human HCC,17 activation of these signaling pathways coincided with suppression of the Ras and JAK/STAT inhibitors RASSF1, RASSF4, SOCS1, SOCS2, SOCS3, and CIS. These inhibitors of the Ras and JAK/STAT signaling pathways, which are frequently inactivated by promoter methylation, have been involved in carcinogenesis.17,19-22,24,25 Epigenetic inactivation of RASSF1 is one of the most common molecular changes in cancer; and a direct correlation between promoter methylation and loss of RASSF1 expression has been shown in many tumor cell lines including hepatoma.17,24 Similarly, the frequency of promoter hypermethylation of SOCS2 has been found to occur in more than 80% of both human HCC and hepatoma cell lines and to correlate with the loss of SOCS2 expression.17 Consistent with these findings, our data demonstrate that the methylation of RASSF1 and SOCS2 promoters is increased in liver tumors from GNMT-knockout mice. Promoter hypermethylation in cancer cells is known to be associated with a variety of alterations in histone modification, including increased H3K27 methylation, which is linked to gene repression.26 Our observation that the level of H3K27me3 bound to RASSF1 and SOCS2 was increased in liver tumors of GNMT-KO mice agrees with this concept. Moreover, our results also indicate that the epigenetic pathways observed in the liver tumors were activated in 3-month-old mice, well in advance of any histological evidence of tumor formation, strongly suggesting a pathogenic relationship between methylation changes and hepatocarcinogenesis.
We have previously demonstrated27,28 that in mice deficient in hepatic SAMe synthesis (methionine adenosyl-transferase 1A knockout mice), SAMe levels in the liver and the SAMe/SAH ratio are reduced and that these animals spontaneously develop steatohepatitis and HCC. Mice deficient in other key enzymes of methionine and folate metabolism, such as cystathionine β synthase and methylenetetrahydrofolate reductase, also have altered levels of hepatic SAMe and a reduced SAMe/SAH ratio and spontaneously develop fatty liver disease.29,30 These findings, together with the present observations using mice deficient in GNMT, suggest that either too much or too little hepatic SAMe in concert with abnormal SAMe/SAH ratios may result in aberrant methylation of DNA and histones, resulting in epigenetic modulation of critical metabolic and carcinogenic pathways in mice, which emphasizes the crucial role of methionine and folate metabolism in maintaining normal liver function.1
Acknowledgments
The authors thank B. Rodríguez for technical assistance, F. Muruzabal for help with the histological preparations, G. Kanel for help with the interpretation of the histology, and OWL Genomics for methionine and SAMe analysis.
Supported by NIH grants AA12677, AA13847, and AT-1576 (to S.C.L. and J.M.M.); DK15289 (to C.W.), PN I+D SAF 2005-00855, HEPADIP-EULSHM-CT-205, and ETORTEK 2005 (to J.M.M. and M.L.M.-C.); Program Ramón y Cajal (to M.L.M.-C.); and Fundación “La Caixa” (to M.L.M.-C., R.M., and A.M.A.).
Abbreviations
- GNMT
glycine N-methyltransferase
- HCC
hepatocellular carcinoma
- H3K27me3
trimethylated lysine 27 in histone 3
- JAK
Janus kinase
- RASSF
Ras-association domain family/tumor suppressor
- SAH
S-adenosylhomocysteine
- SAMe
S-adenosylmethionine
- SOCS
suppressor of cytokine signaling
- STAT
signal transducer and activator of transcription
- WT
wild type
Footnotes
Published online in Wiley InterScience (www.interscience.wiley.com).
Potential conflict of interest: Nothing to report.
References
- 1.Mato JM, Lu SC. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology. 2007;45:1306–1312. doi: 10.1002/hep.21650. [DOI] [PubMed] [Google Scholar]
- 2.Clarke S, Banfield K. S-Adenosylmethionine-dependent methyltransferase. In: Carmel R, Jacobsen DW, editors. Homocysteine in Health and Disease. Cambridge, UK: Cambridge University Press; 2001. pp. 63–78. [Google Scholar]
- 3.Mudd SH, Brosnan JT, Brosnan ME, Jacobs RL, Stabler SP, Allen RH, et al. Methyl balance and transmethylation fluxes in humans. Am J Clin Nutr. 2007;85:19–25. doi: 10.1093/ajcn/85.1.19. [DOI] [PubMed] [Google Scholar]
- 4.Rowling MJ, McMullen MH, Schalinske KL. Vitamin A and its derivatives induce hepatic glycine N-methyltransferase and hypomethylation of DNA in rats. J Nutr. 2002;132:365–369. doi: 10.1093/jn/132.3.365. [DOI] [PubMed] [Google Scholar]
- 5.Ozias M, Schalinske KL. All-trans-retinoic acid rapidly induces glycine N-methyltransferase in a dose-dependent manner and reduces circulating methionine and homocysteine levels in rats. J Nutr. 2003;133:4090–3094. doi: 10.1093/jn/133.12.4090. [DOI] [PubMed] [Google Scholar]
- 6.Luka Z, Capdevila A, Mato JM, Wagner C. A glycine N-methyltransferase knockout mouse model for humans with deficiency of this enzyme. Transgenic Res. 2006;15:393–397. doi: 10.1007/s11248-006-0008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mudd SH, Cerone R, Schiaffino MC, Fantasia AR, Minniti GU, Caruso U, et al. Glycine N-methyltransferase deficiency: a novel inborn error causing persistent isolated hypermethioninaemia. J Inherit Metab Dis. 2001;24:448–464. doi: 10.1023/a:1010577512912. [DOI] [PubMed] [Google Scholar]
- 8.Augoustides-Savvopoulou P, Luka Z, Karyda S, Stabler SP, Allen RH, Patsiaoura K, et al. Glycine N-methyltransferase deficiency: a new patient with a novel mutation. J Inherit Metab Dis. 2003;26:745–749. doi: 10.1023/B:BOLI.0000009978.17777.33. [DOI] [PubMed] [Google Scholar]
- 9.Ogawa H, Fujikoa M. Purification and properties of glycine N-methyltransferase from rat liver. J Biol Chem. 1982;257:3447–3452. [PubMed] [Google Scholar]
- 10.Chen YM, Shiu JY, Tzeng SJ, Shih LS, Chen YL, Lui WY, Chen PH. Characterization of glycine-N-methyltransferase-gene expression in human hepatocellular carcinoma. Int J Cancer. 1998;75:787–793. doi: 10.1002/(sici)1097-0215(19980302)75:5<787::aid-ijc20>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 11.Avila MA, Berasain C, Torres L, Martin-Duce A, Corrales FJ, Yang H, et al. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J Hepatol. 2000;33:907–914. doi: 10.1016/s0168-8278(00)80122-1. [DOI] [PubMed] [Google Scholar]
- 12.Tseng TL, Shih YP, Huang YC, Wang CW, Chen PH, Chang JG, et al. Genotypic and phenotypic characterization of a putative tumor susceptibility gene, GNMT, in liver cancer. Cancer Res. 2003;63:647–654. [PubMed] [Google Scholar]
- 13.Chen SY, Lin JR, Darbba R, Lin P, Liu TY, Chen YM. Glycine N-methyltransferase tumor susceptibility gene in the benzo(a)pyrene-detoxification pathway. Cancer Res. 2004;64:3617–3623. doi: 10.1158/0008-5472.CAN-03-3726. [DOI] [PubMed] [Google Scholar]
- 14.Fraga MF, Ballester E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, et al. High-performance capillary electrophoretic method for the quantification of 5-methyl 2′-deoxycytidine in genomic DNA: application to plant, animal and human cancer tissues. Electrophoresis. 2002;23:1677–1681. doi: 10.1002/1522-2683(200206)23:11<1677::AID-ELPS1677>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 15.Fraga MF, Ballester E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 16.Garcea R, Daino L, Pascales R, Simile MM, Puddu M, Ruggiu ME, et al. Protooncogene methylation and expression in regenerating liver and preneoplastic liver nodules in the rat by diethylnitrosamine: effect of variations of S-adenosylmethionine:S-adenosylhomocysteine ratio. Carcinogenesis. 1989;10:1183–1192. doi: 10.1093/carcin/10.7.1183. [DOI] [PubMed] [Google Scholar]
- 17.Calvisi DF, Ladu S, Gorden A, Farina M, Conner EA, Lee JS, et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130:1117–1128. doi: 10.1053/j.gastro.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 18.Liu S-P, Li Y-S, Chen Y-J, Chiang E-P, Li AF-Y, Lee Y-H, et al. Glycine N-methyltransferase−/− mice develop chronic hepatitis and glycogen storage disease in the liver. Hepatology. 2007;46:1413–1425. doi: 10.1002/hep.21863. [DOI] [PubMed] [Google Scholar]
- 19.Yang B, Guo M, Herman JG, Clark DP. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am J Pathol. 2003;163:1101–1107. doi: 10.1016/S0002-9440(10)63469-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okochi O, Hibi K, Sakai M, Inoue S, Takeda S, Kaneko T, et al. Methylation-mediated silencing of SOCS-1 gene in hepatocellular carcinoma derived from cirrhosis. Clin Cancer Res. 2003;9:5295–5298. [PubMed] [Google Scholar]
- 21.Eckfeld K, Hesson L, Vos MD, Bieche I, Latif F, Clark GJ. RASSF4/AD037 is a potential ras effector/tumor suppressor of the RASSF family. Cancer Res. 2004;64:8688–8693. doi: 10.1158/0008-5472.CAN-04-2065. [DOI] [PubMed] [Google Scholar]
- 22.Ogata H, Kobayashi T, Chinen T, Takaki H, Sanada T, Minoda Y, et al. Deletion of the SOCS3 gene in liver parenchymal cells promotes hepatitis-induced hepatocarcinogenesis. Gastroenterology. 2006;131:317–319. doi: 10.1053/j.gastro.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 23.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 24.Agathanggelou A, Cooper WN, Latuf F. Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res. 2005;65:3497–3508. doi: 10.1158/0008-5472.CAN-04-4088. [DOI] [PubMed] [Google Scholar]
- 25.Tomasi S, Damman R, Zhang Z, Wang Y, Liu L, Tsark WM, et al. Tumor susceptibility of Rassf1a knockout mice. Cancer Res. 2005;65:92–98. [PubMed] [Google Scholar]
- 26.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Genet. 2007;8:286–298. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 27.Lu SC, Alvarez L, Huang ZZ, Chen L, An W, Corrales FJ, et al. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc Natl Acad Sci U S A. 2001;98:5560–5565. doi: 10.1073/pnas.091016398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martínez-Chantar ML, Corrales FJ, Martínez-Cruz A, García-Trevijano ER, Huang ZZ, Chen L, et al. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1A. FASEB J. 2002;16:1292–1294. doi: 10.1096/fj.02-0078fje. [DOI] [PubMed] [Google Scholar]
- 29.Robert K, Nehmé J, Bourdon E, Pivert G, Friguet B, Delcayre C, et al. Cystathionine β synthase deficiency promotes oxidative stress, fibrosis, and steatosis in mice liver. Gastroenterology. 2005;128:1408–1415. doi: 10.1053/j.gastro.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 30.Schwahn BC, Chen Z, Laryea MD, Wendel U, Lussier-Cacan S, Genest J, Jr, et al. Homocysteine-betaine interactions in a murine model of 5,10-methylenetetrahydrofolate reductase deficiency. FASEB J. 2003;17:512–514. doi: 10.1096/fj.02-0456fje. [DOI] [PubMed] [Google Scholar]