

Abstract
Blastic transformation of chronic myelogenous leukemia (CML) is characterized by the presence of nonrandom, secondary genetic abnormalities in the majority of Philadelphia1 clones, and loss of p53 tumor suppressor gene function is a consistent finding in 25–30% of CML blast crisis patients. To test whether the functional loss of p53 plays a direct role in the transition of chronic phase to blast crisis, bone marrow cells from p53+/+ or p53−/− mice were infected with a retrovirus carrying either the wild-type BCR/ABL or the inactive kinase-deficient mutant, and were assessed for colony-forming ability. Infection of p53−/− marrow cells with wild-type BCR/ABL, but not with the kinase-deficient mutant, enhanced formation of hematopoietic colonies and induced growth factor independence at high frequency, as compared with p53+/+ marrow cells. These effects were suppressed when p53−/− marrow cells were coinfected with BCR/ABL and wild-type p53. p53-deficient BCR/ABL-infected marrow cells had a proliferative advantage, as reflected by an increase in the fraction of S+G2 phase cells and a decrease in the number of apoptotic cells. Immunophenotyping and morphological analysis revealed that BCR/ABL-positive p53−/− cells were much less differentiated than their BCR/ABL-positive p53+/+ counterparts. Injection of immunodeficient mice with BCR/ABL-positive p53−/− cells produced a transplantable, highly aggressive, poorly differentiated acute myelogenous leukemia. In marked contrast, the disease process in mice injected with BCR/ABL-positive p53+/+ marrow cells was characterized by cell infiltrates with a more differentiated phenotype and was significantly retarded, as indicated by a much longer survival of leukemic mice. Together, these findings directly demonstrate that loss of p53 function plays an important role in blast transformation in CML.
Keywords: oncogene, tumor suppressor gene, leukemia
Chronic myelogenous leukemia (CML) is a clonal disorder arising from neoplastic transformation of the hematopoietic stem cell (1). The typical clinical course of CML involves progression from a protracted chronic phase to an accelerated phase with a duration of 1–1.5 years followed by a rapidly fatal blastic phase (1). In the chronic phase, the disease process is characterized by increased numbers of immature myeloid precursors in bone marrow, peripheral blood, spleen, and liver. Myeloid precursors retain the ability to terminally differentiate so that the hallmark of the disease is a marked granulocytosis. Spleen and liver are also sites of extramedullary hematopoiesis that contributes to the prominent hepatosplenomegaly typically found in CML patients. The transition from chronic phase to blast crisis is characterized by a dramatic increase in the number of blast cells in hematopoietic tissues. Such cells exhibit proliferative advantage over normal cells and loss of differentiation potential. Cytogenetically, CML is characterized by a t(9;22) translocation (2) in which sequences of the c-ABL and the breakpoint cluster region (BCR) genes on chromosomes 9 and 22, respectively, join to form the BCR/ABL fusion genes of the Philadelphia chromosome (Ph). Such genes encode the p210bcr/abl oncogenic tyrosine kinase (3), which plays an essential role in the pathogenesis of CML (4, 5).
The molecular mechanisms involved in the evolution from chronic to acute leukemia remain poorly understood. The frequent and nonrandom cytogenetic changes detected during disease progression in 80% of patients suggest that superimposed secondary genetic events account for the evolution to blast crisis (6). However, there is no direct evidence that the malfunction of any specific gene induces the progression from chronic phase to blast crisis. One of the most frequent alterations is the isochromosome 17 (i17) (6), which results in the loss of the short arm of chromosome 17 to which the p53 tumor suppressor gene has been mapped (7). Structural alterations (e.g., deletions, rearrangements, point mutations) of p53 are exceedingly rare in CML chronic phase, but are detected in 25–30% of CML blast crisis, suggesting the involvement of p53 in the progression of the disease (8).
To determine whether inactivation of the p53 tumor suppressor gene plays an essential role in the transition of CML chronic phase to blast crisis, we analyzed the influence of BCR/ABL on the growth characteristics of bone marrow cells (BMCs) derived from p53 knockout mice (9). p53−/− BMCs infected with a retrovirus carrying the BCR/ABL oncogene, but not the tyrosine-kinase deficient mutant, were differentiation-arrested, had a low apoptotic index, and exhibited a higher colony-forming ability in the presence or absence of interleukin 3 (IL-3), as compared with p53+/+ BMCs. When injected into immunodeficient mice, BCR/ABL-infected p53−/− marrow cells generated a rapidly fatal, transplantable, acute leukemia highly reminiscent of the disease process in humans.
MATERIALS AND METHODS
Preparation of Viral Stocks.
The pSRαMSVtkneo vector carrying either the p210BCR/ABL(b3/a2) wild-type (WT) sequence or the 1172R kinase-negative p210BCR/ABL(b3/a2) mutant under the control of the long terminal repeat of the murine sarcoma virus and the NEO resistance gene under the herpes simplex thymidine kinase promoter (10), and the pSV-Ψ−-E-MLV packaging vector were obtained from C. Sawyers (University of California, Los Angeles). Helper-free retroviral stocks were prepared by transient hyperexpression in COS-7 cells as described (11). Stocks were collected in Iscove’s modified Dulbecco’s medium (IMDM) supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 50 units/ml penicillin, 50 μg/ml streptomycin, and 50 μM 2-mercaptoethanol, filtered through 0.45-μm pore-size filters, and stored at −80°C. The viral titers were quantitated by infection of 105 Rat-2 cells. At 72 hr postinfection, BCR/ABL genome transfer was assessed by expression of BCR/ABL protein. Viral stocks were adjusted to give approximately the same level of protein expression for BCR/ABL WT and its 1172R mutant.
The cDNA of human WT p53 was obtained from B. Vogelstein (The Johns Hopkins University School of Medicine, Baltimore), and was cloned into the BamHI site of the LXSN vector. The expression of p53 was under the control of the long terminal repeat of the Moloney murine leukemia virus, while expression of the NEO resistance gene was driven by the simian virus 40 early promoter. The construct was electroporated into the GP + E86 packaging cell line (12), and virus-producing cells were obtained after a 2- to 3-week selection in G418-containing medium (1 mg/ml). Viral stock-containing supernatants were collected 3 days after monolayer formation. Titers of the stocks were determined by infection of Rat-2 cells and scoring the number of G418-resistant colonies.
Mice.
p53−/− (C57BL/6TacfBR-[KO]p53N4) and p53+/+ mice were obtained from Taconic Farms. C57BL/6-SCID-SzJ mice were from The Jackson Laboratory. The genetic background of the latter animals is identical to that of C57BL/6 mice, except for the severe combined immunodeficiency (SCID) mutation.
Retroviral Infections.
BMCs were infected as described (13). Twenty-four hours later, cells were washed three times and used immediately or cultured for 14 days in IMDM supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 50 units/ml penicillin, 50 μg/ml streptomycin, and 10 units/ml of each of the following recombinant growth factors (all from Genetics Institute, Cambridge, MA): murine IL-3, murine Kit ligand, and human IL-6. G418 (1.6 mg/ml) was added to the culture to select a pure population of infected cells. Uninfected p53−/− cells died at this G418 concentration, despite bearing a NEO gene within each p53 allele, in accordance with Strasser et al. (14).
In Vitro Analyses of the Cells.
Clonogenic assays were performed as described (15). Cells were plated in MethoCult H4230 semisolid medium (Stem Cell Technologies, Vancouver) in the presence of the indicated concentrations of recombinant murine IL-3 and colonies were scored from days 9 to 12.
The threshold concentration of IL-3 was established for each batch of the cytokine and corresponds to the lowest concentration able to support colony formation. Cell-cycle distribution was analyzed as described (16). Apoptosis was examined using the TACS 1 Klenow in situ apoptosis detection kit (Trevigen, Gaithersburg, MD), according to the manufacturer’s protocol.
Protein expression was analyzed as described (17), except that the filters were blotted simultaneously with murine monoclonal antibodies anti-p53 (Ab-3; Oncogene Science) and anti-ABL (clone 8E9; the kind gift of R. Arlinghaus, M.D. Anderson Medical Center, Houston).
Monoclonal antibodies were obtained from PharMingen, except for the anti-murine CD34 (L. Lasky, Genentech), and all were used as described (18) to determine the phenotype of BMCs after retroviral infection. Cytospins of cultured BMCs were routinely stained with Giemsa stain and occasionally stained with Sudan Black B and nonspecific esterase to detect myelomonocytic and purely monocytic differentiation, respectively.
In Vivo Leukemogenesis.
Because of the mixed genetic background of the test cells, 106 bone marrow cells were injected i.v. into total body-irradiated (350 rads) C57BL-SCID-SzJ immunodeficient mice, 1 or 14 days after infection. The genotype of the recipient mice is similar to that of the test cells (C57BL ≫ 129/Sv). The presence of BCR/ABL transcripts in peripheral blood cells was tested by reverse transcripase–PCR (RT-PCR) as described (17, 18). For pathological examination, tissue sections from various organs were fixed in phosphate-buffered formalin and embedded in paraffin. Slides were routinely stained with hematoxylin/eosin (H&E) and, in addition, with stain for chloroacetate esterase (Leder staining) to detect differentiated myeloid cells.
RESULTS
Absence of Functional p53 Affects BMC Colony Formation After BCR/ABL Infection.
To determine whether the absence of an intact p53 gene affects BCR/ABL-induced transformation, BMCs from p53−/− and WT p53+/+ mice were infected with the insert-less or the BCR/ABL-containing virus, cultured for 14 days in the presence of G418, plated in semisolid medium, and assessed for colony formation. In our experimental conditions, p53 status did not affect colony formation of BMCs infected with the insert-less virus; in the presence of a threshold concentration of IL-3, the BCR/ABL-carrying virus increased colony formation by 3- to 4-fold in p53-expressing cells (p53+/+), whereas colony formation of p53 nonexpressing cells (p53−/−) was increased 14- to 16-fold (Table 1). When cells were plated in methylcellulose in the absence of IL-3, BCR/ABL induced 4- to 7-fold more colonies from p53−/− than from p53+/+ BMCs, and these differences were even more evident in secondary colony formation assays, in which BCR/ABL stimulated about 10-fold more colonies from p53−/− cells than from p53+/+ cells (Table 1). Colony-forming assays performed with marrow cells plated 1 day after infection gave similar results, but with a lower frequency of colonies (not shown). These differences in colony formation were not due to a higher susceptibility of p53−/− marrow cells to retroviral infection, as indicated by a similar number of proviral integrants in wild-type and p53−/− marrow cells 72 h after infection with supernatant from GP+E86 packaging cells producing the LXSN virus (Fig. 1). Morphologic analysis revealed mainly large and dense colonies derived from p53−/− cells infected with BCR/ABL, whereas infection of p53+/+ cells produced colonies of various sizes, mostly spread, and only a few that were large and dense (not shown).
Table 1.
Effect of p53 expression on the number of colonies derived from BMCs after infection with BCR/ABL
| Virus | Cells | Primary
culture
|
Secondary culture
|
||
|---|---|---|---|---|---|
| IL-3 + | IL-3 − | IL-3 + | IL-3 − | ||
| E | +/+ | 23 ± 7 | 0 | 13 ± 3 | — |
| E | −/− | 27 ± 10 | 0 | 20 ± 6 | — |
| BCR/ABL | +/+ | 89 ± 17 | 4 ± 1 | 86 ± 12 | 2 ± 1 |
| BCR/ABL | −/− | 376 ± 68 | 28 ± 2 | 887 ± 154 | 26 ± 8 |
| BCR/ABL[1172] | +/+ | 32 ± 15 | 0 | 11 ± 4 | — |
| BCR/ABL[1172] | −/− | 34 ± 9 | 0 | 16 ± 3 | — |
| E + p53[WT] | −/− | 24 ± 3 | 0 | 15 ± 7 | — |
| BCR/ABL + p53[WT] | −/− | 74 ± 10 | 3 ± 2 | 81 ± 7 | 1 ± 2 |
BMCs from p53+/+ or p53−/− mice were infected with BCR/ABL, BCR/ABL[1172R] mutant, p53 wild-type (p53[WT]), or insert-less (E) retroviral constructs. After 14 days in liquid culture with G418, 105 cells were plated in G418-containing semisolid medium for 12 days (primary colonies) in the presence or absence of threshold concentrations (0.1 unit/ml) of recombinant murine IL-3. For secondary colony assays, 10 large colonies from each group growing in the absence of IL-3 were plucked individually and 50 cells from each colony were plated. From colonies grown in the presence of IL-3, 104 cells were replated in threshold concentrations of IL-3 in the presence of G418. Colonies were counted 9 to 12 days later. Results represent mean ± SD from 3–4 experiments.
Figure 1.
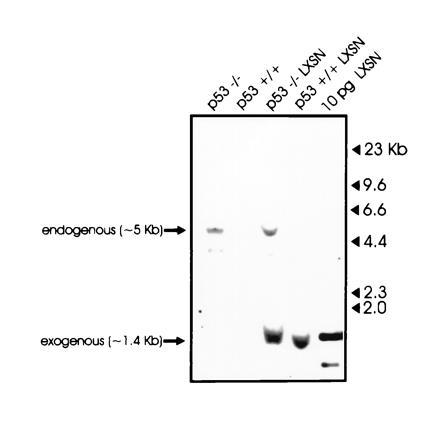
Integration of the LXSN vector in infected p53+/+ or p53−/− marrow cells. Genomic DNA (30 μg) from p53+/+ or p53−/− marrow cells, infected or not with the LXSN retroviral vector, was digested with BamHI/NheI restriction endonucleases, electrophoresed, and probed with a 32P-labeled 1.4-kb BamHI/NheI fragment corresponding to the NEO cassette of the LXSN retroviral vector. As a positive control, 10 pg of the LXSN vector was also digested with BamHI/NheI restriction enzymes.
The virus containing the kinase-negative BCR/ABL [1172R] mutant did not induce any of the changes described above (Table 1), indicating that BCR/ABL tyrosine kinase activity, which is essential for BCR/ABL-mediated leukemogenesis (10), is also necessary for induction of increased colony formation from p53-deficient BMCs.
Expression of BCR/ABL proteins in p53+/+ and p53−/− BMCs was documented by SDS/PAGE followed by Western blotting with specific monoclonal antibodies (Fig. 2A). Single-colony RT-PCR revealed expression of BCR/ABL transcripts in all of the 20 colonies tested after selection in the presence of G418 (not shown).
Figure 2.
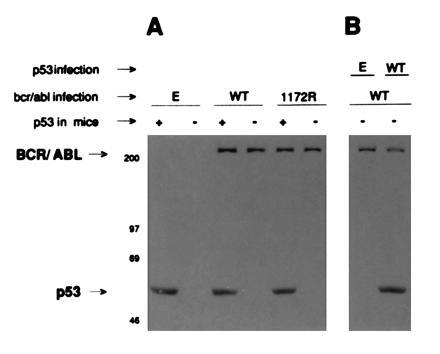
(A) Expression of BCR/ABL in infected BMCs. BMCs from p53+/+ (+) or p53−/− (−) mice were infected with insert-less (E), wild-type BCR/ABL (WT), or 1172R BCR/ABL virus (1172R). (B) Expression of BCR/ABL and p53 proteins in bone marrow coinfected cells. BMCs from p53−/− (−) mice were coinfected with WT BCR/ABL (WT) and also p53 wild-type (WT) or insert-less virus (E). p210BCR/ABL and p53 proteins were detected by SDS/PAGE followed by Western blotting.
To confirm that the increased colony-forming ability of p53−/− cells infected with BCR/ABL was due to lack of p53 expression, and not to unknown differences between p53+/+ and p53−/− cells, we attempted to restore p53 expression in p53−/− cells by infection with a virus containing the human WT p53 cDNA. Enforced expression of the WT p53 reduced BCR/ABL-driven colony formation of p53−/− cells to the level observed with p53+/+ cells (Table 1). The efficiency of retroviral infection was determined by SDS/PAGE and Western blotting detection of p210BCR/ABL and/or p53 proteins (Fig. 2B). In addition, BCR/ABL and WT p53 were simultaneously detected in 80% of individual colonies from p53−/− coinfected cells, as indicated by RT-PCR phenotyping (data not shown). Infection of p53−/− cells with p53 transactivation-deficient mutant did not reduce BCR/ABL-induced colony formation (not shown).
Effect of p53 on Cell-Cycle Status of Marrow Cells Expressing BCR/ABL.
After 14 days of culture in the presence of G418 and a threshold concentration of IL-3, almost all p53+/+ and p53−/− cells infected with insert-less virus resided in the G1 phase of the cell cycle (98 ± 1% and 96 ± 1%, respectively), and only a few of them were in S and G2/M phases (2 ± 1% and 4 ± 1% respectively). Introduction of BCR/ABL had only a modest effect on the percentage of p53+/+ cells in S and G2 phases. This fraction dramatically increased (41%) in p53−/− marrow cells, consistent with the ability of BCR/ABL expression to stimulate hematopoietic cell proliferation in the absence of p53.
Use of an apoptosis-detection kit revealed that BCR/ABL-positive p53−/− cells contained the lowest percentage of apoptotic cells (5 ± 2%) in comparison with BCR/ABL-positive p53+/+ cells (17 ± 4%), p53−/− cells (26 ± 4%), and p53+/+ cells (61 ± 8%).
Overall, BCR/ABL-positive p53−/− cells had a much higher proliferative potential and a much lower apoptotic index, as compared with BCR/ABL-positive p53+/+ cells.
p53 Status Affects Immunophenotype and Morphology of BCR/ABL-Infected Cells.
The immunophenotype of p53+/+ and p53−/− BMCs infected with the insert-less virus was similar to that of p53+/+ BCR/ABL-infected BMCs, except that BCR/ABL increased the percentage of CD34+ cells by about 2-fold. In contrast, expression of BCR/ABL in p53−/− cells increased the percentage of CD34+ cells by about 4-fold and decreased the percentage of cells expressing lineage-specific differentiation markers (myelomonocytic GR-1 and Mac-1, erythroid TER-119, lymphoid CD3 and B220), indicating that BCR/ABL-positive p53−/− cells were less differentiated of the BCR/ABL-positive p53+/+ counterparts. The immunophenotypic data are consistent with the expansion of a population of BCR/ABL-positive p53−/− early progenitor cells with a limited potential to undergo terminal differentiation.
Morphologic analysis of these cells (Fig. 3) reflected the immunophenotypic differences. BCR/ABL-positive p53+/+ cells showed definitive maturation along the myeloid lineage to fully mature granulocytes (Fig. 3A). Occasionally, differentiation toward macrophages was also noted. In contrast, BCR/ABL-positive p53−/− cells consisted of a much less differentiated myeloid population with many cells representing intermediate forms (promyelocytes and myelocytes) (Fig. 3B). A moderate degree of cellular dysplasia was also present in these cells, as indicated by cytoplasmic hypergranulation, nuclear enlargement, and hyperchromasia.
Figure 3.
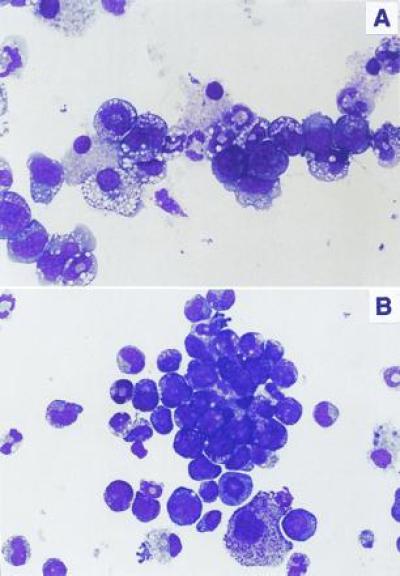
Morphology of BCR/ABL-infected BMCs depends on p53 status. Cells were stained with Giemsa stain. (A) p53+/+ cells. (B) p53−/− cells. (×600.)
BCR/ABL-Positive p53−/− Cells Induce a CML-Blast Crisis-Like Syndrome in Vivo.
To assess the leukemogenic potential of BCR/ABL-positive p53−/− and BCR/ABL-positive p53+/+ cells, 106 marrow cells (1 or 14 days after infection) were injected i.v. into irradiated immunodeficient SCID-SzJ mice; they were chosen because their genetic background is very similar to that of the test cells. As an indicator of disease progression, 4 weeks after injection of the test cells, BCR/ABL transcripts were detected by RT-PCR in the peripheral blood of mice inoculated with BCR/ABL-positive p53−/− cells, but not in mice injected with the BCR/ABL-positive p53+/+ cells (Fig. 4). BCR/ABL transcripts were detected only after 16 weeks in mice injected with BCR/ABL-positive p53+/+ cells (Fig. 4). The level of detection of our RT-PCR procedure was of 10 BCR/ABL-positive cells in a mixture of 106 normal marrow cells (data not shown).
Figure 4.
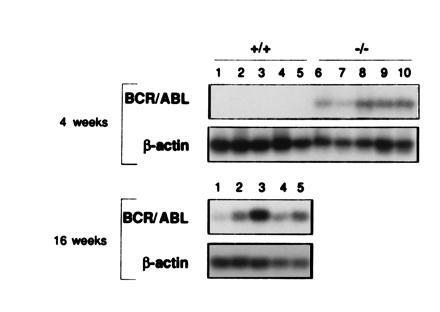
Development of BCR/ABL-induced leukemia depends on p53 status. BCR/ABL transcripts detected by RT-PCR followed by Southern blotting in peripheral blood of immunodeficient mice 4 weeks or 16 weeks after injection of 14-day in vitro cultured BCR/ABL-infected p53+/+ cells (lanes 1–5) or BCR/ABL-infected p53−/− cells (lanes 6–10), respectively. No bands were detected from RNA samples subjected to PCR amplification only. β-Actin served as a control of the integrity of the RNA used for RT-PCR.
Mice injected i.v. with 106 BCR/ABL-positive p53−/− cells died within 7–9 weeks; in marked contrast, mice inoculated with BCR/ABL-positive p53+/+ cells were apparently disease-free up to 20 weeks (Fig. 5); thereafter, most of them became sick (increased abdominal size, reduced food intake, lethargy) and eventually succumbed to the disease, as revealed by necropsy.
Figure 5.
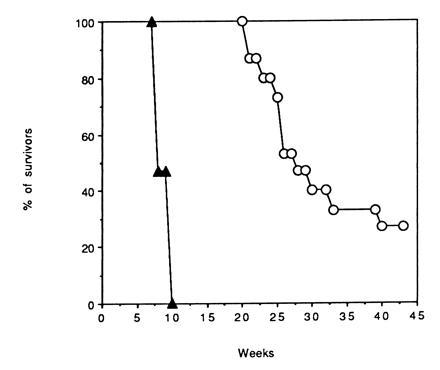
Influence of p53 phenotype on the survival of mice carrying BCR/ABL-infected BMCs. Total body-irradiated SCID-SzJ mice (15 per group, two different experiments) were injected i.v. with 106 p53+/+ (○) or p53−/− (▴) BMCs infected with the BCR/ABL-containing virus. In each group, eight and seven mice were injected with cells maintained in culture in the presence of Kit ligand, IL-3, and IL-6 for 1 or 14 days (in the presence of G418) after retroviral infection, respectively. Mice died of leukemia as confirmed at necropsy. Mice (five per group) injected with p53+/+ or p53−/− BMCs infected with the insert-less virus were disease-free during the period of observation.
The peripheral blood white cell count of the mice injected with BCR/ABL-infected p53−/− marrow cells was very high, ranging from 382 to 612 × 103 cells per μl. Analysis of various organs from these mice revealed markedly enlarged spleens, with a diffuse cellular infiltrate replacing both red and white pulp (Fig. 6A). Higher magnification (Fig. 6B) identified most of the cells as blasts based on their large nuclei with fine chromatin and prominent nucleoli. A limited degree of myeloid differentiation was noted. This finding is highlighted by the red staining with chloroacetate esterase, which is highly specific for cells of the myeloid lineage, of up to 5% of the cells (Fig. 6C). These findings are diagnostic of acute myelogenous leukemia without maturation. Similar leukemic infiltrates were seen in the bone marrow, the liver, the lung, and, focally, in the kidney (not shown). Marrow showed areas of diffuse necrosis as sometimes seen in acute, highly aggressive leukemias. Only a few leukemic cells (<1%) from bone marrow stained with Sudan Black B and nonspecific esterase. This finding is also consistent with a low degree of myeloid differentiation.
Figure 6.
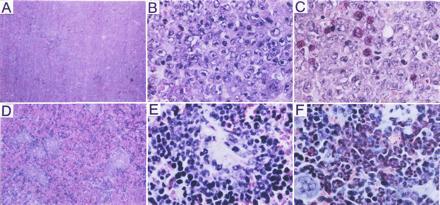
Pathologic analysis of the spleens of mice injected with BCR/ABL-infected p53−/− (A–C) or p53+/+ (D–F) marrow cells. (A) Spleen of mouse injected with BCR/ABL-infected p53−/− cells (H&E stain; ×25). (B) High-power image of splenic leukemic infiltrate (H&E stain; ×350). (C) Chloroacetate esterase stain (red) to detect myeloid differentiation (Leder staining; ×350). (D) Spleen of mouse injected with BCR/ABL-infected p53+/+ marrow cells. (E) High power image of splenic leukemic infiltrate (H&E stain; ×350). (F) As in C (Leder staining; ×350).
The peripheral blood white cell count of SCID mice inoculated with BCR/ABL-infected p53+/+ cells was increased above normal values and ranged between 57 and 119 × 103 cells per μl. Spleens from these mice were moderately enlarged and contained multiple foci of a dense cellular infiltrate obliterating the white and, to a lesser degree, the red pulp (Fig. 6D). At higher magnification, most of the cells represented myeloid elements at various stages of differentiation including large clusters of fully mature granulocytes (Fig. 6E). Occasionally, scattered megakaryocytes were also detected. Chloroacetate esterase (Leder) stain was positive in >50% of all cells and in >75% of cells without megakaryocytic differentiation (Fig. 6F). Bone marrow from these mice showed myeloid hyperplasia with ≈80% of the cells positive for Sudan Black B and <5% for nonspecific esterase staining (not shown). Thus, in contrast to the BCR/ABL-positive p53−/− cells that produced acute myelogenous leukemia mimicking blast transformation in CML, injection of BCR/ABL-positive p53+/+ cells led to myeloid cell hyperplasia resembling chronic phase of CML.
DISCUSSION
CML is an attractive model to study the molecular basis of disease progression because it evolves in clinically and temporally distinct stages. The best documented genetic change, the Philadelphia chromosome translocation, involves hematopoietic stem cells or their most immediate progeny. Thus, the clonal nature of CML makes it possible to investigate the pathogenetic role of the additional genetic alterations that mark the evolution of the disease process.
Disruption of p53 function by deletion, rearrangement, or point mutation is found in 25–30% of CML blast crisis patients, but is exceedingly rare during the chronic phase (19). Such loss of function is consistently found in CML blast crisis with a myeloid, but not a lymphoid, phenotype (8). This contrasts with the approximately equal frequency of p53 mutations in de novo acute myelogenous and lymphocytic leukemias (8). Interestingly, p53 knock-out mice, which are susceptible to developing a variety of tumors including lymphoma, do not manifest leukemic transformation in the myeloid or erythroid lineage (9, 20).
Bone marrow progenitor cells infected with a retrovirus carrying the BCR/ABL gene form morphologically and immunophenotypically distinguishable colonies at higher frequency than mock-infected cells (13). Consistent with those findings, BCR/ABL-infected normal marrow cells exhibited a limited proliferative advantage as reflected in the higher propensity to form hematopoietic colonies in methylcellulose (Table 1). After 14 days in liquid culture supplemented with IL-3, these cells had morphological and immunophenotypic features of terminal differentiation. Most of these cells appeared to reside in the G0/G1 phase of the cell cycle and a significant fraction had hypodiploid DNA content, consistent with the presence of apoptotic cells. In marked contrast, BCR/ABL-infected p53-deficient marrow cells had a much higher proliferative potential, reflected in exuberant primary and secondary colony formation and in the emergence, at higher frequency, of growth factor-independent colony-forming cells. DNA content analysis was consistent with the proliferative advantage of these cells, as reflected in the high fraction of S+G2 phase cells and the low apoptotic index. Similar results were obtained if p53+/− marrow cells were coinfected with a p53 transactivation-deficient mutant and BCR/ABL (not shown).
The combined effects of BCR/ABL expression and loss of p53 function result in the precocious emergence of a cell phenotype to which each gene contributes individually. p53 plays a pivotal role in the regulation of cell cycle progression and the commitment to apoptosis (21). BCR/ABL promotes growth factor independence (22) and prolongs cell survival, perhaps by a p53-independent mechanism (23). It is also known that Myc synergizes with and is required for BCR/ABL mediated transformation (24), and it induces cell proliferation, but not apoptosis in mouse fibroblasts lacking p53 (25). Thus, its activation may explain, at least in part, the phenotype of BCR/ABL-positive p53−/− marrow cells. The morphology of BCR/ABL-infected p53-deficient marrow cells was strikingly different from that of wild-type BCR/ABL-infected cells; most of the p53-deficient cells were blast-like and only few exhibited a moderate degree of differentiation. As expected, immunophenotypic analysis of these cells revealed few that expressed late differentiation markers of myeloid, erythroid, and lymphoid lineages, whereas the frequency of cells expressing the stem/progenitor CD34 antigen was significantly higher. These data are consistent with the expansion of a population of early progenitor cells that are unable to complete the differentiation program.
This phenotype is reminiscent of that induced by constitutive expression of v-abl or p210bcr/abl in growth factor-dependent cell lines, in which both growth factor independence and inability to differentiate in response to granulocyte colony-stimulating factor are observed (22, 26). The differentiation arrest of p53-deficient BCR/ABL-infected marrow cells might be a direct consequence of the loss of a differentiation-inducing function ascribed to WT p53. Overexpression of p53 has been shown to promote differentiation in hematopoietic cells (27). Such an effect might be due to a direct transactivation of regulators of the cell differentiation program; however, it is also possible that the p53-induced G1 arrest is sufficient to trigger differentiation in cells committed to enter such a pathway.
Loss of p53 function in marrow cells might recapitulate the genetic or epigenetic changes that lead to indefinite maintenance in culture of growth factor-dependent lines. This interpretation is consistent with the recent demonstration that the absence of p53 allows the direct immortalization of fetal liver hematopoietic cells by a retroviral construct carrying the myc and raf oncogenes (28).
The most compelling evidence for the leukemogenic potential of BCR/ABL-infected p53-deficient marrow cells is their ability to induce leukemia when injected in mice. The disease process was rapidly fatal (7- to 9-week survival) and, at necropsy, hematopoietic tissues showed an abundance of highly undifferentiated myeloid blast cells. Nonhematopoietic organs such as lungs, liver, and kidneys were also involved. The myeloid phenotype of the leukemic cells arising from BCR/ABL-positive p53-deficient marrow cells might reflect a higher susceptibility of myeloid progenitors to undergo leukemic transformation in the absence of p53 expression, consistent with p53 loss of function in myeloid but not in lymphoid blast crisis (8). However, it remains also possible that the emergence of blast cells with a myeloid phenotype has been favored by the exposure of hematopoietic progenitors to myeloid-specific growth factors at the time of retroviral infection. Upon introduction of blast cells isolated from the spleen of an affected mouse into secondary recipients, the acute leukemia was even more aggressive and mice died of the disease in 3–4 weeks (data not shown). By comparison, the disease process induced by injection of BCR/ABL-infected, WT p53 marrow cells was significantly retarded, morphologically resembling chronic phase CML, consistent with the observations previously described by others (4, 5). The rapidly fatal acute leukemia in mice injected with BCR/ABL-infected p53-deficient marrow cells is highly reminiscent of blast crisis in CML patients. Disruption of p53 function is, however, only observed in 25–30% of blast crisis cases; since there is no evidence to suggest that this subgroup presents with clinically distinctive features, it seems likely that other genetic abnormalities detected in blast crisis have similar pathogenetic effects in marrow progenitor cells “initiated” by BCR/ABL in the pathway leading to a more malignant phenotype.
Mice transplanted with leukemic cells originating from BCR/ABL-infected, p53-deficient marrow cells exhibited a more aggressive disease process characterized by disruption of the architecture of virtually every organ by massive leukemic cell infiltrates, and by an early death. Perhaps, loss of p53 function leads to secondary genetic changes resulting in the emergence of leukemic clones with a more malignant phenotype. This interpretation is consistent with recent studies indicating that the increased propensity of serially passaged p53-deficient astrocytes to form tumors correlated with the occurrence of dramatic alterations in ploidy and karyotype (29).
In conclusion, the model of CML blast crisis described here might prove useful for dissecting genetic changes associated with the development of a highly malignant phenotype, and for assessing the pathogenetic role of other abnormalities found in CML blast crisis. Finally, this model might also be useful in testing novel therapeutic strategies.
Acknowledgments
We thank B. Perussia, W. E. Mercer, and E. Canaani for critical reading of the manuscript. We thank D. Dicker for flow cytometry analysis. This study was supported by National Institutes of Health and American Cancer Society grants to B.C.
Footnotes
Abbreviations: CML, chronic myelogenous leukemia; BMCs, bone marrow cells; BCR, breakpoint cluster region; ABL, Abelson oncogene; IL-3, interleukin 3; WT, wild-type; SCID, severe combined immunodeficiency; RT-PCR, reverse transcriptase–PCR.
References
- 1.Kantarjan H M, Deisseroth A, Kurzrok R, Estrov Z, Talpaz M. Blood. 1993;82:691–703. [PubMed] [Google Scholar]
- 2.de Klein A, van Kessel A G, Grosveld G, Bartram C R, Hagemeijer A, Bootsma D, Spurr N K, Heisterkamp N, Groffen J, Stephenson J. Nature (London) 1982;300:765–767. doi: 10.1038/300765a0. [DOI] [PubMed] [Google Scholar]
- 3.Konopka J B, Watanabe S M, Witte O N. Cell. 1984;37:1035–1042. doi: 10.1016/0092-8674(84)90438-0. [DOI] [PubMed] [Google Scholar]
- 4.Daley G R, Van Etten R A, Baltimore D. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 5.Kelliher M A, McLaughlin J, Witte O N, Rosenberg N. Proc Natl Acad Sci USA. 1990;87:6649–6653. doi: 10.1073/pnas.87.17.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rowley J D, Testa J R. Adv Cancer Res. 1982;36:103–148. doi: 10.1016/s0065-230x(08)60423-6. [DOI] [PubMed] [Google Scholar]
- 7.Miller C, Mohandas T, Wolf D, Prokocimer M, Rotter V, Koeffler P M. Nature (London) 1986;319:783–785. doi: 10.1038/319783a0. [DOI] [PubMed] [Google Scholar]
- 8.Prokocimer M, Rotter V. Blood. 1994;8:2391–2411. [PubMed] [Google Scholar]
- 9.Donehower L A, Harvey M, Slagle B L, McArthur M J, Montgomery C A, Butel J S, Bradley A. Nature (London) 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 10.Pendergast A M, Gishizky M L, Havlik M H, Witte O N. Science. 1993;247:1079–1083. [Google Scholar]
- 11.Muller A J, Young J C, Pendergast A M, Pondel M, Landau N R, Littman D R, Witte O N. Mol Cell Biol. 1991;11:1785–1792. doi: 10.1128/mcb.11.4.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Markowitz D, Goff S, Bank A. J Virol. 1988;62:1120–1124. doi: 10.1128/jvi.62.4.1120-1124.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gishizki M L, Witte O N. Science. 1992;256:836–839. doi: 10.1126/science.1375394. [DOI] [PubMed] [Google Scholar]
- 14.Strasser A, Harris A W, Jacks T, Cory S. Cell. 1994;79:329–339. doi: 10.1016/0092-8674(94)90201-1. [DOI] [PubMed] [Google Scholar]
- 15.Skorski T, Nieborowska-Skorska M, Campbell K, Iozzo R V, Zon G, Darzynkiewicz Z, Calabretta B. J Exp Med. 1995;182:1–9. doi: 10.1084/jem.182.6.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skorski T, Nieborowska-Skorska M, Nicolaides N C, Szczylik C, Iversen P, Iozzo R V, Zon G, Calabretta B. Proc Natl Acad Sci USA. 1994;81:4504–4508. doi: 10.1073/pnas.91.10.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skorski T, Nieborowska-Skorska M, Barletta C, Malaguarnera L, Szczylik C, Chen S T, Lange B, Calabretta B. J Clin Invest. 1993;92:194–202. doi: 10.1172/JCI116549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skorski T, Szczylik C, Ratajczak M Z, Malaguarnera L, Gewirtz A M, Calabretta B. J Exp Med. 1992;175:743–750. doi: 10.1084/jem.175.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feinstein E, Cimino G, Gale R P, Alimena G, Berthier R, Kishi K, Goldman J, Zaccaria A, Berrebi A, Canaani E. Proc Natl Acad Sci USA. 1991;88:6293–6297. doi: 10.1073/pnas.88.14.6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacks T, Remington L, Williams B O, Schmitt E M, Halachmi S, Bronson R T, Weinberg R. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 21.Oren M. Semin Cancer Biol. 1994;5:221–227. [PubMed] [Google Scholar]
- 22.Laneuville P, Heisterkamp N, Groffen J. Oncogene. 1991;6:275–282. [PubMed] [Google Scholar]
- 23.Sanchez-Garcia I, Grütz G. Proc Natl Acad Sci USA. 1995;92:5287–5291. doi: 10.1073/pnas.92.12.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sawyers C L, Callahan W, Witte O N. Cell. 1992;70:901–910. doi: 10.1016/0092-8674(92)90241-4. [DOI] [PubMed] [Google Scholar]
- 25.Hermeking H, Eick D. Science. 1994;265:2091–2093. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- 26.Rovera G, Valtieri M, Mavilio F, Reddy E P. Oncogene. 1987;1:29–35. [PubMed] [Google Scholar]
- 27.Feinstein E, Gale R P, Reed J, Canaani E. Oncogene. 1992;7:1853–1857. [PubMed] [Google Scholar]
- 28.Metz T, Harris A W, Adams J M. Cell. 1995;82:29–36. doi: 10.1016/0092-8674(95)90049-7. [DOI] [PubMed] [Google Scholar]
- 29.Yahanda A M, Bruner J M, Donehower L A, Morrison R S. Mol Cell Biol. 1995;15:4249–4259. doi: 10.1128/mcb.15.8.4249. [DOI] [PMC free article] [PubMed] [Google Scholar]