

Abstract
Variations in OPRK1, which encodes the κ-opioid receptor, are associated with the risk for alcohol dependence. Sequencing DNAs with higher and lower risk haplotypes revealed an insertion/deletion (indel) with a net addition of 830 bp located 1986 bp upstream of the translation start site (1389 bp upstream of the transcription start site). We demonstrated that the upstream region extending from −1647 to −10 bp or from −2312 to −10 bp (relative to the translation start site) could function as a promoter in transient transfection assays. We then determined that the presence of the indel reduced transcriptional activity by half. We used a PCR assay to genotype individuals in 219 multiplex alcohol-dependent families of European American descent for the presence or absence of this indel. Family-based association analyses detected significant evidence of association of this insertion with alcoholism; the longer allele (with the indel), which had lower expression, is associated with higher risk for alcoholism. This indel is, therefore, a functional regulatory variation likely to explain at least part of the association of OPRK1 with alcohol dependence.
INTRODUCTION
Opioids regulate many brain functions relevant to addictions, including reward, reinforcement, mood and psychomotor stimulation (1–5). In particular, stimulation of the κ-opioid receptor (KOR) reduces the release of dopamine in the nucleus accumbens of the mouse and generates aversive states (1,2). The effects of stimulating or inhibiting the κ-opioid system are complex and may be affected by species, strain, duration of exposure and drinking paradigm (6). Stimulating the KOR with a selective KOR agonist reduces voluntary ethanol intake in rats in a two-bottle choice paradigm (7), but chronic treatment potentiated ethanol intake during the alcohol deprivation effect in rats that had long-term exposure to ethanol (3). Blocking the KOR in mice that normally drink ethanol (C57/BL6) with a KOR-specific antagonist, nor-binaltorphimine, increases alcohol self-administration (8). Walker and Koob (9) reported that nor-binaltorphimine selectively reduced ethanol self-administration in rats made dependent on ethanol but not in non-dependent rats. Mice in which the KOR has been disrupted drink half as much ethanol as either wild-type or heterozygous mice (10). Ethanol treatment, particularly in combination with cocaine, reduces KOR mRNA in the VTA of rats (11). Acute ethanol increased dopamine levels in the nucleus accumbens to a greater extent in KOR−/− mice than in wild-type mice, and nor-binaltorphimine increased ethanol-evoked dopamine levels (12). Chronic ethanol treatment led to increased dynorphin B in the nucleus accumbens which persisted at least 21 days after ceasing treatment (13). The κ-opioid system also appears to be involved in ethanol withdrawal-related seizures (14–16).
A recent family-based study in European Americans demonstrated that variations in OPRK1, the gene encoding the KOR, and in PDYN, which encodes its dynorphin ligand, were associated with alcohol dependence (17). The associated SNPs in OPRK1 were in intron 2 of the gene; two coding SNPs were not associated (17). The association of OPRK1 with alcoholism was confirmed by a second study, reporting that a haplotype of OPRK1 was associated with alcohol dependence (18), although individual SNPs were not; it should be noted that Zhang et al. (18) did not test any SNPs in intron 2, the region where association had earlier been reported. An earlier study of three coding SNPs in Taiwanese Han subjects found no association between OPRK1 and alcohol dependence (19). The finding that non-coding SNPs, but not coding SNPs, were associated with alcohol dependence suggests that the association is with regulatory variation in the gene. Non-coding OPRK1 variations have also been associated with opioid addiction (20), as was the coding SNP G36T (21). The association of variants in PDYN with alcohol dependence was also replicated (22). Variations in other opioid receptor genes were not associated with alcohol dependence in the same large, family-based sample in which the association with OPRK1 was reported (17). The involvement of genes encoding both an opioid receptor (OPRK1) and its ligand (PDYN) makes biological sense, indicating that variations in signaling through this system, by varying either the ligand or receptor, affect alcohol consumption and alcohol dependence.
OPRK1 is located on human chromosome 8q11.2. There are four exons within the 22 kb gene (RefSeq NM_000912); the 5′ exon is non-coding (21,23). When we found that non-coding variations in OPRK1 were associated with alcohol dependence (17), we sequenced DNA from individuals with different haplotypes to identify additional variations. We identified a previously unknown insertion/deletion (indel) and examined its effect on the regulation of gene expression. We also tested whether that indel was associated with alcoholism.
RESULTS
Polymorphisms in the OPRK1 promoter region
Sequencing the exons and proximal 5′ region of OPRK1 in 16 human samples (half carrying the lower risk and half carrying the higher risk haplotype) led to the detection of six new SNPs (reported to dbSNP: rs35970029, rs34418807, rs35991105, rs34709943, rs35373196, rs35160174). At all of the SNPs, the lower risk samples were homozygous for the reference sequence, whereas some of the higher risk samples were heterozygous. We also detected a new complex indel involving the deletion of 11 bp from −1975 to −1985 (all positions are relative to the reference genome sequence NT_008183.18 and the translation start site of OPRK1, RefSeq NM_000912) and the insertion of 841 bp at −1986 bp relative to the reference genome sequence, for a net insertion of 830 bp (reported to dbSNP: rs35566036). All eight of the samples with the lower risk haplotype lacked the indel and matched the reference genome sequence. All the eight of the samples with the higher risk haplotype had a copy of the indel. A diagram of the gene, based upon the reference sequence, is shown in Figure 1. The minor allele containing the indel is ancestral; BLAST analysis shows that the indel is contained in the Chimp genome (NW_001240342.1) and in the genome of the Rhesus macaque (AC202275.5).
Figure 1.

5′ region of the OPRK1 gene and fragments used in promoter assays. Boxes 1 and 2 are exons; gray, 5′ untranslated sequence; black, coding sequence. Transcription in direction of top arrow. 1.6 kb is the fragment extending from −1647 to −10 bp relative to the translation start site; 2.3 kb is the fragment with the reference sequence, from −2312 to −10 bp; 3.1 kb is the fragment with the same ends but containing the additional 830 bp of the indel (minor allele).
Effects of indel on gene expression
To determine whether the presence of the indel affected transcription in vitro, transient transfection assays were carried out. First, the promoter region extending approximately −1.6 kb upstream of the translation start site was compared with that extending to −2.3 kb on the reference sequence, in DNA without the indel (Fig. 2). The additional 665 bp had a small but significant negative effect upon gene expression, reducing it to 87% that of the shorter fragment (P < 0.01). Next, the construct with the indel (3.1) was compared with its counterpart without the indel (2.3). The presence of the indel lowered transcription activity to 53% of that in its absence (P < 10−11).
Figure 2.

Effect of the indel on promoter function in vitro. Black bars represent the mean expression, gray bars are ±1 standard error. 1.6 kb is the fragment extending from −1647 to −10 bp relative to the translation start site; 2.3 kb is the fragment with the reference sequence, from −2312 to −10 bp; 3.1 kb is the fragment from −2312 to −10 bp but containing the additional 830 bp indel.
Association of the indel with alcohol dependence
The strong effect of the indel on gene expression, and the finding that it was common to eight individuals with the higher risk haplotype of OPRK1, led us to examine its potential association with alcoholism. We genotyped 1914 individuals in 219 European American families in which at least three first-degree relatives were alcohol dependent. Genotyping was by PCR amplification followed by size determination of the fragments on agarose gels (Fig. 3). The reference sequence was the major allele. The allele with the indel (the net 830 bp insertion) was the minor allele, with a frequency of 28% in these families; genotypes were in Hardy–Weinberg equilibrium. The indel was in high LD (as measured by D′) with all of the SNPs we had previously genotyped (17) (data not shown). However, owing to differences in allele frequencies, the indel had only a low correlation (r2) with many of the SNPs (Fig. 4).
Figure 3.
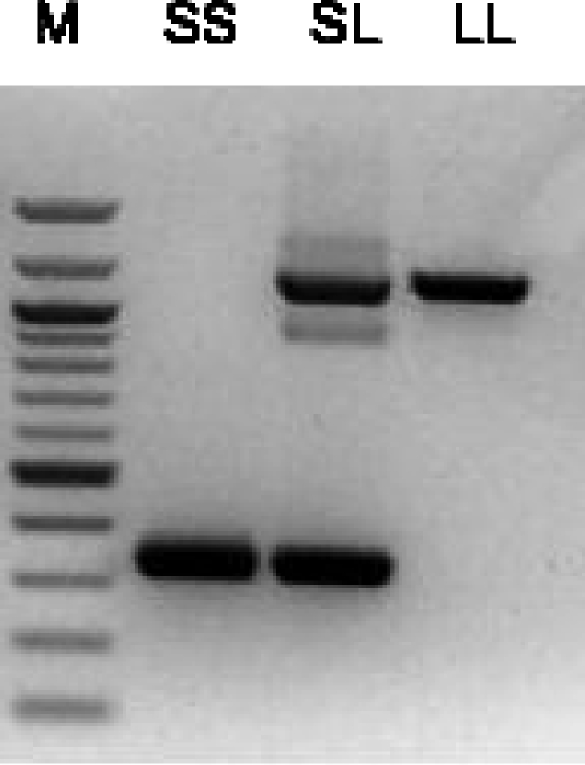
Genotyping of the indel. PCR was carried out with primers HE3236 and HE3244 as described in the text, followed by electrophoresis on 1% agarose gels. A homozygote lacking the indel is shown as SS, a homozygote with the indel as LL and a heterozygote as SL. M is a 100 bp DNA ladder (New England Biolabs, Ipswitch, MA, USA); the darker bands are 500 and 1000 bp.
Figure 4.

Linkage disequilibrium among genotyped markers (r2). At top is a diagram of the gene showing the relative position of the oprk1ins indel (rs35566036) and of the SNPs previously reported (17); associated SNPs are boxed. Below is a diagram of LD between SNPs, calculated as r2.
Family-based association analysis by the Pedigree Disequilibrium Test (PDT) demonstrated that the indel was associated with alcohol dependence (P = 0.01). The minor allele, with the net additional 830 bp, was overtransmitted to affected individuals (265 transmissions versus 235 non-transmissions) and overtransmitted to the affected sibling in a discordant sibling pair (495 transmissions to the affected sibling; 453 transmissions to the unaffected sibling). Reanalyzing the data after coding as ‘unknown’ individuals who were not alcohol dependent but were dependent on other illicit drugs (in case there was shared genetic vulnerability at this locus) yielded a similar result (P = 0.02). Comparing the pattern of association results previously reported for SNPs (17) with that of LD as measured by r2 (Fig. 4), we found that four of the five SNPs for which r2 with the indel was >35% were significant and the fifth was marginal; in contrast, only one of the eight SNPs for which r2 with the indel was lower (<35%) was significant and one was marginal.
DISCUSSION
We previously reported that SNPs in intron 2 of OPRK1, the gene encoding the KOR, were associated with alcohol dependence; three synonymous SNPs were not associated (17). To better understand this association, we sequenced DNA from individuals with higher risk and lower risk alleles to detect additional genetic variations. Sequencing revealed several novel SNPs along with an indel upstream of the translation start site. This indel, at −1986, added a net 830 bp of sequence and was present in the sequenced individuals who were alcohol dependent. The indel was in high LD with most of the SNPs that we had previously reported associated with alcohol dependence (17). Here we demonstrate that the 5′ region of OPRK1 can function as a promoter in vitro. We then analyzed the effect of the indel on gene expression. Presence of this indel reduced transcription by nearly half. Genotyping and family-based analysis of our data set of multiplex alcohol-dependent families demonstrated that this indel was significantly associated with alcoholism. Therefore, we conclude that this indel is a functional variation that explains at least part of the association of OPRK1 with alcoholism.
The κ-opioid system has been implicated in several effects of ethanol. The effects on ethanol consumption of modulating the κ-opioid system are complex and differ in different species, strains and conditions (6). Stimulation of the KOR reduces voluntary ethanol drinking in rats (7). Blocking the KOR increases voluntary ethanol intake in C57/BL6 mice (8). Although the phenotype of voluntary drinking of ethanol is clearly not the same as alcohol dependence, these results are consistent with our results, which demonstrate that the presence of the indel that reduced gene expression in vitro by half was associated with a higher risk for alcoholism. In a different system, a selective KOR antagonist reduced ethanol self-administration in rats that had been made dependent on ethanol, but not on control rats (9). Mice homozygous for a disruption in Oprk1 drink less ethanol in a two-bottle choice paradigm than either wild-type or heterozygous mice (10), however, suggesting that complete deletion of the KOR reduces drinking in mice, but a 50% reduction in KOR levels may not affect it. Again, alcohol dependence in humans is a much more complex phenotype.
Our study demonstrates that an indel 1986 bp upstream of the translational start site in OPRK1 reduces promoter activity by about half and is associated with alcohol dependence. These data represent the demonstration of a functional variant in OPRK1 that is associated with alcoholism.
MATERIALS AND METHODS
Subjects
Subjects were members of families in which at least three first-degree relatives were alcohol dependent [meeting both DSM-IIIR lifetime criteria for alcohol dependence (24) and Feighner criteria for definite alcohol dependence (25)]. Only non-Hispanic families of European American descent (219 families) were analyzed. The ascertainment and assessment have previously been described (26,27) and are available in detail at zork.wustl.edu/niaaa/coga_instruments/resources.html. A genetically informative subset of families was selected as described in more detail by Foroud et al. (27). Subjects were collected at six centers in the USA: Indiana University, State University of New York Health Science Center, University of Connecticut, University of Iowa, University of California/San Diego and Washington University, St Louis. The institutional review boards of all participating institutions approved the study. Probands were identified through alcoholism treatment programs, and after providing written informed consent, probands and their relatives were administered a validated poly-diagnostic instrument, the Semi-Structured Assessment for the Genetics of Alcoholism (SSAGA) interview (28,29).
DNA sequencing
The OPRK1 5′ region and first exon, from −2312 (numbering here is relative to the translational start site ATG in RefSeq NM_000912) to +453, were amplified from 16 human genomic DNAs, eight from individuals with the higher risk haplotype and eight with the lower risk haplotype previously described (17). Primers (Table 1) were designed on the basis of the NCBI reference genome contig (NT_008183.18). We used the primer pairs shown in Table 2 to amplify the region from −2312 to +452 in four overlapping fragments. PCR products were directly sequenced using the ABI BigDye Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and separated on an ABI 3100 Genetic Analyzer (Applied Biosystems). Sequences were aligned using MacVector (MacVector Inc., Cary, NC, USA), and polymorphic sites were checked on the electropherograms. New sequences were submitted to dbSNP (SNPs: rs35970029, rs34418807, rs35991105, rs34709943, rs35373196, rs35160174; indel rs35566036).
Table 1.
Primer sequences
| Primer | Sequence |
|---|---|
| HE3026 | CATAGTGTTTGCATTGAGTAGAATG |
| HE3027 | AATTTCCCCAGTGGATTCAC |
| HE3028 | GGACATTAGCTGTGGGATGC |
| HE3029 | TGCACCTGGATTTTCATATTG |
| HE3040 | AGACGGAATGGAAGAGAACG |
| HE3041 | AGAACTGCACCTCGAAAGCC |
| HE3032 | TGTCTCTGGGAACCATAGGTAAG |
| HE3033 | TCTCAGCACTCCACCAAAAAC |
| HE3058 | CCGGTCGACCATAGTGTTTGCATTGAGTAGAATG |
| HE3059 | CCGGTCGACGGACATTAGCTGTGGGATGC |
| HE3061 | CCCGGTACCATTGCAGCAGGAAGGCGAGGA |
| HE3236 | GAGGGCTTCTCAATGCTCTG |
| HE3244 | GGATTTGACCACCAGCGTC |
Table 2.
PCR primer pairs used for DNA sequencing and size of the fragments amplified
| Primer pair | Positiona | Length |
|---|---|---|
| HE3026/HE3027 | −2312 to −1577 | 736 (no indel); 1566 (with indel) |
| HE3028/HE3029 | −1647 to −928 | 720 |
| HE3040/HE3041 | −1118 to −381 | 738 |
| HE3032/HE3033 | −428 to 453 | 881 |
aPosition relative to the translational start site of OPRK1 (NM_000912) on the NCBI reference genome (NT_008183.18).
Cloning of OPRK1 upstream regions
Two human genomic DNAs were chosen as templates to amplify the OPRK1 upstream regions, one containing the indel (6006) and one lacking it (5003). The primer pairs are listed in Table 3; we added a SalI site at the 5′ end and a KpnI site at the 3′ end of the fragments to facilitate cloning into the luciferase reporter plasmid pXP2 (30). Amplified fragments were purified, digested with SalI and KpnI and cloned into pXP2 that was similarly digested. Identity of the fragments was confirmed by restriction mapping and DNA sequencing.
Table 3.
OPRK1 constructs for transfection assays
| Name | Positiona | Primer pairs | DNAb | Length (bp) |
|---|---|---|---|---|
| OPRK-1.6 | −1647 to −10 | HE3059/HE3061 | 5003 | 1638 |
| OPRK-2.3 | −2312 to –10 | HE3058/HE3061 | 5003 | 2303 |
| OPRK-3.1 | −2312 to –10c | HE3058/HE3061 | 6006c | 3133c |
Fragments noted were cloned into pXP2 for assays of promoter activity.
aPosition relative to the translational start site of OPRK1 (NM_000912) on the NCBI reference genome (NT_008183.18).
bIndividual genomic DNA used as template.
cIndividual 6006 has DNA with the indel; amplification using the same primers produced a fragment with the additional 830 bp indel.
Transient transfection assays
HepG2 cells were grown in six-well plates (Corning, NY, USA) in MEM with 10% fetal bovine serum (FBS; Life Technologies, Rockville, MD, USA) and 2 mm glutamine. Cells (3.6 × 105 cells per well) were washed with 2 ml of medium without FBS and then transfected using Fugene 6 (Roche Diagnostics, Indianapolis, IN, USA) with a total of 3.3 µg DNA (2 µg test plasmid, 0.13 µg pCMV-β-galactosidase as an internal control and 1.2 µg pUC19 DNA) for 5 h. Complete medium (2 ml) containing FBS was then added and incubation was continued for 25 h. Cells were washed with 1× phosphate-buffered saline and resuspended in 100 µl of 1× reporter lysis buffer (Promega, Madison, WI, USA). Cells were lysed by three freeze–thaw cycles, followed by vortex mixing for 15 s. Twenty microliters of each lysate was assayed for luciferase activity using the Luciferase Assay System (Promega), with activity measured for 60 s on an Lmax Plate Luminometer (Molecular Devices, Sunnyvale, CA, USA). Five microliters of each lysate was assayed for β-galactosidase activity for 30 s using the Galacto-Light™ Plus System (Applied Biosystems, Benford, MA, USA) and the Lmax Plate Luminometer.
In each of four independent experiments, the test constructs and external positive control (pXP2 with an unrelated promoter for normalization) and negative control (pXP2 without a promoter fragment) were transfected in triplicate. Luciferase activity/β-galactosidase activity was calculated for each sample and then normalized to the average of the three external controls in the same experiment, with the average value of the external controls set to 100. Data are presented as mean relative luciferase activity ±1 standard error of the mean. Significance was determined by a t-test (two-tailed, unequal variance; Microsoft Excel). Negative controls averaged 0.8% of the positive control.
Genotyping
Allele frequencies for the indel were determined by PCR amplification of 30 ng of genomic DNA using HE3026/HE3027 (Table 1; Fig. 1); this yields a 736 bp fragment in the reference genomic sequence (NT_008183.18) or a 1566 bp fragment if the indel is present. HotStar Taq (Qiagen, Valencia, CA, USA) was used to minimize background. The final reaction contained 1× PCR buffer, 200 µM of each dNTP, 3.5 mm Mg2+, 1× Q-solution, 0.4 µM primer, 1.5 unit of HotStarTaq DNA polymerase (Qiagen) in 15 µl total volume. PCR reactions were carried out in GeneAmp PCR system 9700 (Applied Biosystems, CA, USA) using the following conditions: 95°C 15 min to activate the HotStarTaq polymerase, then 35 cycles of 95°C for 20 s, 60°C for 15 s, 72°C for 2 min 45 s, final extension at 72°C for 10 min. Aliquots of the PCR products (6 µl) were separated on 1% agarose gels (E-Gel 96, Invitrogen, CA, USA). Each gel was read independently by two people, and the data were combined by a third individual.
There were dropouts from the initial genotyping, so fresh aliquots of DNA from those individuals were analyzed using a different primer pair, HE3236/HE3244 (Table 1), which yielded a 336 bp band (without indel) or a 1166 bp band with the indel; we reasoned that reducing the size of the fragments should make the assay more robust to potential DNA degradation. The final reaction contained 1× R-Taq Master Mix (Bulleye R-Taq DNA pol 2.0× Mix, MidWest Scientific, St Louis, MO, USA), 0.375 µM primer in 20 µl total volume with 15 ng genomic DNA. PCR reactions were carried out in GeneAmp PCR system 9700 (Applied Biosystems) using the following conditions: 95°C for 3 min for initial DNA denaturation, then 35 cycles of 94°C for 20 s, 62°C for 15 s, 72°C for 1 min 30 s, final extension at 72°C for 7 min. Aliquots (6 µl) of PCR product were separated on 2% agarose gels (E-Gel 96, Invitrogen) and independently read by three individuals. Only unambiguous data were used. Results of the genotyping were tested for Mendelian inheritance using the program PEDCHECK (75). Marker allele frequency and heterozygosity were computed using the program USERM13 (76).
Statistical analyses
Family-based association analyses were performed using PDT (78) as implemented in the program UNPHASED (version 2.404) (31). Individuals were considered affected if they were alcohol dependent on the basis of meeting both DSM-IIIR criteria for alcohol dependence (24) and Feighner criteria for definite alcoholism (25); there were 876 affected and 915 unaffected by this definition. Because of the possibility of shared genetic vulnerability between dependence on alcohol and dependence on other illicit drugs, a secondary analysis was done in which all individuals who were alcohol dependent were considered affected (876), individuals not dependent on either alcohol or illicit drugs were considered unaffected (835), and individuals who were not alcohol dependent but were dependent on illicit drugs were considered unknown (203). We report results using the PDTaverage option, which weighs each family equally in computing the overall test statistic. Linkage disequilibrium between the indel and SNPs previously genotyped in these families (17) was evaluated using HAPLOVIEW (32).
FUNDING
This national collaborative study is supported by the NIH Grant U10AA008401 from the National Institute on Alcohol Abuse and Alcoholism (NIAAA) and the National Institute on Drug Abuse (NIDA). Genotyping was carried out using the facilities of the Center for Medical Genomics at Indiana University School of Medicine, which is supported in part by the Indiana Genomics Initiative of Indiana University (INGEN®); INGEN is supported in part by The Lilly Endowment, Inc.
Supplementary Material
ACKNOWLEDGEMENTS
The Collaborative Study on the Genetics of Alcoholism (COGA)—Co-Principal Investigators: B. Porjesz, V. Hesselbrock, H. Edenberg, L. Bierut—includes nine different centers where data collection, analysis and storage take place. The nine sites and Principal Investigators and Co-Investigators are University of Connecticut (V. Hesselbrock); Indiana University (H.J. Edenberg, J. Nurnberger Jr, T. Foroud); University of Iowa (S. Kuperman, R. Crowe); SUNY Downstate (B. Porjesz); Washington University in St Louis (L. Bierut, A. Goate, J. Rice); University of California at San Diego (M. Schuckit); Howard University (R. Taylor); Rutgers University (J. Tischfield); Southwest Foundation (L. Almasy). Zhaoxia Ren serves as the NIAAA Staff Collaborator. This work is in memory of Henri Begleiter and Theodore Reich, Principal and Co-Principal Investigators of COGA since its inception; we are indebted to their leadership in the establishment and nurturing of COGA and acknowledge with great admiration their seminal scientific contributions to the field. Funding to pay the Open Access publication charges for this article was provided by NIH Grant U10AA008401.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Spanagel R., Herz A., Shippenberg T.S. Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc. Natl Acad. Sci. USA. 1992;89:2046–2050. doi: 10.1073/pnas.89.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spanagel R., Herz A., Shippenberg T.S. The effects of opioid peptides on dopamine release in the nucleus accumbens: an in vivo microdialysis study. J. Neurochem. 1990;55:1734–1740. doi: 10.1111/j.1471-4159.1990.tb04963.x. [DOI] [PubMed] [Google Scholar]
- 3.Holter S.M., Henniger M.S., Lipkowski A.W., Spanagel R. Kappa-opioid receptors and relapse-like drinking in long-term ethanol-experienced rats. Psychopharmacology. 2000;153:93–102. doi: 10.1007/s002130000601. [DOI] [PubMed] [Google Scholar]
- 4.Vaccarino A.L., Olson G.A., Olson R.D., Kastin A.J. Endogenous opiates: 1998. Peptides. 1999;20:1527–1574. doi: 10.1016/s0196-9781(99)00166-7. [DOI] [PubMed] [Google Scholar]
- 5.Kreek M.J., Bart G., Lilly C., LaForge K.S., Nielsen D.A. Pharmacogenetics and human molecular genetics of opiate and cocaine addictions and their treatments. Pharmacol. Rev. 2005;57:1–26. doi: 10.1124/pr.57.1.1. [DOI] [PubMed] [Google Scholar]
- 6.Shippenberg T.S., Zapata A., Chefer V.I. Dynorphin and the pathophysiology of drug addiction. Pharmacol. Ther. 2007;116:306–321. doi: 10.1016/j.pharmthera.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindholm S., Werme M., Brene S., Franck J. The selective kappa-opioid receptor agonist U50,488H attenuates voluntary ethanol intake in the rat. Behav. Brain Res. 2001;120:137–146. doi: 10.1016/s0166-4328(00)00368-5. [DOI] [PubMed] [Google Scholar]
- 8.Shippenberg T.S., Chefer V.I., Zapata A., Heidbreder C.A. Modulation of the behavioral and neurochemical effects of psychostimulants by kappa-opioid receptor systems. Ann. NY Acad. Sci. 2001;937:50–73. doi: 10.1111/j.1749-6632.2001.tb03558.x. [DOI] [PubMed] [Google Scholar]
- 9.Walker B.M., Koob G.F. Pharmacological evidence for a motivational role of kappa-opioid systems in ethanol dependence. Neuropsychopharmacology. 2008;33:643–652. doi: 10.1038/sj.npp.1301438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovacs K.M., Szakall I., O'Brien D., Wang R., Vinod K.Y., Saito M., Simonin F., Kieffer B.L., Vadasz C. Decreased oral self-administration of alcohol in kappa-opioid receptor knock-out mice. Alcohol. Clin. Exp. Res. 2005;29:730–738. doi: 10.1097/01.alc.0000164361.62346.d6. [DOI] [PubMed] [Google Scholar]
- 11.Rosin A., Lindholm S., Franck J., Georgieva J. Downregulation of kappa opioid receptor mRNA levels by chronic ethanol and repetitive cocaine in rat ventral tegmentum and nucleus accumbens. Neurosci. Lett. 1999;275:1–4. doi: 10.1016/s0304-3940(99)00675-8. [DOI] [PubMed] [Google Scholar]
- 12.Zapata A., Shippenberg T.S. Endogenous kappa opioid receptor systems modulate the responsiveness of mesoaccumbal dopamine neurons to ethanol. Alcohol. Clin. Exp. Res. 2006;30:592–597. doi: 10.1111/j.1530-0277.2006.00069.x. [DOI] [PubMed] [Google Scholar]
- 13.Lindholm S., Ploj K., Franck J., Nylander I. Repeated ethanol administration induces short- and long-term changes in enkephalin and dynorphin tissue concentrations in rat brain. Alcohol. 2000;22:165–171. doi: 10.1016/s0741-8329(00)00118-x. [DOI] [PubMed] [Google Scholar]
- 14.Beadles-Bohling A.S., Wiren K.M. Anticonvulsive effects of kappa-opioid receptor modulation in an animal model of ethanol withdrawal. Genes Brain Behav. 2006;5:483–496. doi: 10.1111/j.1601-183X.2005.00200.x. [DOI] [PubMed] [Google Scholar]
- 15.Beadles-Bohling A.S., Wiren K.M. Alteration of kappa-opioid receptor system expression in distinct brain regions of a genetic model of enhanced ethanol withdrawal severity. Brain Res. 2005;1046:77–89. doi: 10.1016/j.brainres.2005.03.043. [DOI] [PubMed] [Google Scholar]
- 16.Beadles-Bohling A.S., Crabbe J.C., Wiren K.M. Elevated prodynorphin expression associated with ethanol withdrawal convulsions. Neurochem. Int. 2000;37:463–472. doi: 10.1016/s0197-0186(00)00056-5. [DOI] [PubMed] [Google Scholar]
- 17.Xuei X., Dick D., Flury-Wetherill L., Tian H.J., Agrawal A., Bierut L., Goate A., Bucholz K., Schuckit M., Nurnberger J., Jr, et al. Association of the kappa-opioid system with alcohol dependence. Mol. Psychiatry. 2006;11:1016–1024. doi: 10.1038/sj.mp.4001882. [DOI] [PubMed] [Google Scholar]
- 18.Zhang H., Kranzler H.R., Yang B.Z., Luo X., Gelernter J. The OPRD1 and OPRK1 loci in alcohol or drug dependence: OPRD1 variation modulates substance dependence risk. Mol. Psychiatry. 2007 doi: 10.1038/sj.mp.4002035. PMID: 17622222; epub ahead of print July 10, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loh el W., Fann C.S., Chang Y.T., Chang C.J., Cheng A.T. Endogenous opioid receptor genes and alcohol dependence among Taiwanese Han. Alcohol. Clin. Exp. Res. 2004;28:15–19. doi: 10.1097/01.ALC.0000106303.41755.B8. [DOI] [PubMed] [Google Scholar]
- 20.Gerra G., Leonardi C., Cortese E., D'Amore A., Lucchini A., Strepparola G., Serio G., Farina G., Magnelli F., Zaimovic A., et al. Human Kappa opioid receptor gene (OPRK1) polymorphism is associated with opiate addiction. Am. J. Med. Genet. B Neuropsychiatry Genet. 2007;144B:771–775. doi: 10.1002/ajmg.b.30510. [DOI] [PubMed] [Google Scholar]
- 21.Yuferov V., Fussell D., LaForge K.S., Nielsen D.A., Gordon D., Ho A., Leal S.M., Ott J., Kreek M.J. Redefinition of the human kappa opioid receptor gene (OPRK1) structure and association of haplotypes with opiate addiction. Pharmacogenetics. 2004;14:793–804. doi: 10.1097/00008571-200412000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams T.J., LaForge K.S., Gordon D., Bart G., Kellogg S., Ott J., Kreek M.J. Prodynorphin gene promoter repeat associated with cocaine/alcohol codependence. Addict. Biol. 2007;12:496–502. doi: 10.1111/j.1369-1600.2007.00069.x. [DOI] [PubMed] [Google Scholar]
- 23.Simonin F., Gaveriaux-Ruff C., Befort K., Matthes H., Lannes B., Micheletti G., Mattei M.G., Charron G., Bloch B., Kieffer B. kappa-Opioid receptor in humans: cDNA and genomic cloning, chromosomal assignment, functional expression, pharmacology, and expression pattern in the central nervous system. Proc. Natl Acad. Sci. USA. 1995;92:7006–7010. doi: 10.1073/pnas.92.15.7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 3rd edn (revised) Washington, DC: American Psychiatric Association Press; 1987. [Google Scholar]
- 25.Feighner J.P., Robins E., Guze S.B., Woodruff R.A., Winokur G., Munoz R. Diagnostic criteria for use in psychiatric research. Arch. Gen. Psychiatry. 1972;26:57–63. doi: 10.1001/archpsyc.1972.01750190059011. [DOI] [PubMed] [Google Scholar]
- 26.Reich T., Edenberg H.J., Goate A., Williams J.T., Rice J.P., Van Eerdewegh P., Foroud T., Hesselbrock V., Schuckit M.A., Bucholz K., et al. Genome-wide search for genes affecting the risk for alcohol dependence. Am. J. Med. Genet. 1998;81:207–215. [PubMed] [Google Scholar]
- 27.Foroud T., Edenberg H.J., Goate A., Rice J., Flury L., Koller D.L., Bierut L.J., Conneally P.M., Nurnberger J.I., Bucholz K.K., et al. Alcoholism susceptibility loci: confirmation studies in a replicate sample and further mapping. Alcohol. Clin. Exp. Res. 2000;24:933–945. [PubMed] [Google Scholar]
- 28.Bucholz K.K., Cadoret R., Cloninger C.R., Dinwiddie S.H., Hesselbrock V.M., Nurnberger J.I.J., Reich T., Schmidt I., Schuckit M.A. A new semi-structured psychiatric interview for use in genetic linkage studies: a report of the reliability of the SSAGA. J. Stud. Alcohol. 1994;55:149–158. doi: 10.15288/jsa.1994.55.149. [DOI] [PubMed] [Google Scholar]
- 29.Hesselbrock M., Easton C., Bucholz K.K., Schuckit M., Hesselbrock V. A validity study of the SSAGA—a comparison with the SCAN. Addiction. 1999;94:1361–1370. doi: 10.1046/j.1360-0443.1999.94913618.x. [DOI] [PubMed] [Google Scholar]
- 30.Nordeen S.K. Luciferase reporter gene vectors for analysis of promoters and enhancers. Biotechniques. 1988;6:454–456. [PubMed] [Google Scholar]
- 31.Dudbridge F. Pedigree disequilibrium tests for multilocus haplotypes. Genet. Epidemiol. 2003;25:115–121. doi: 10.1002/gepi.10252. [DOI] [PubMed] [Google Scholar]
- 32.Barrett J.C., Fry B., Maller J., Daly M.J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.