

Abstract
Clinical studies demonstrate a positive correlation between the extent of matrix metalloproteinase (MMP) activation and malignant progression of precancerous lesions. Therefore, identification of effective, well-tolerated MMP inhibitors represents a rational chemopreventive strategy. A variety of agents, including proteinases and thiol-oxidizing compounds, activate MMPs by initiating release of the propeptide's cysteine sulfur “blockage” of the MMP active site. Despite the importance of the propeptide's cysteine thiol in preserving MMP latency, limited studies have evaluated the effects of reduced thiols on MMP function. This study investigated the effects of two naturally occurring nonprotein thiols, i.e., glutathione (GSH) and N-acetylcysteine (NAC), on activation, function, and cellular–extracellular matrix interactions of the basement-membrane-degrading gelatinase, MMP-9. Our results reveal that NAC and GSH employ protein S-thiolation to inhibit organomercurial activation of pro-MMP-9. Gelatinase activity assays showed that GSH and NAC significantly inhibited MMP-9 but not MMP-2 function, implying isoform structural specificity. Immunoblot analyses, which suggested GSH interacts with MMP-9's active-site Zn, were corroborated by computational molecular modeling. Cell invasion assays revealed that NAC enhanced endostatin's ability to inhibit human cancer cell invasion. Collectively, these data demonstrate that nonprotein thiols suppress MMP-9 activation and function and introduce the prospect for their use in chemopreventive applications.
Keywords: Glutathione, Nonprotein thiols, MMPs, Cell invasion, Thiolation, Chemoprevention, Free radicals
Both physiological processes such as angiogenesis and pathologic events like tumor cell invasion are dependent on proteolytic degradation of the extracellular matrix (ECM) by enzymes that include matrix metalloproteinases (MMPs) [1]. In physiological states, MMP activity is closely regulated at three distinct levels, i.e., gene expression, enzyme activation, and enzyme inhibition [2,3]. Conversely, it has been proposed that loss of MMP regulation, by prolonged MMP activation and/or concurrent inactivation of tissue inhibitors of MMPs (TIMPs), contributes to diseases, including cancer [2,3]. Consistent with their putative carcinogenesis-facilitating roles, increased MMP activity has been detected in a variety of human cancers, including colorectal, breast, lung, prostate, and head and neck squamous cell carcinoma (HNSCC) [4-8]. Studies conducted on HNSCC resected tumors revealed that MMP functional increases were apparent in tumor as well as in inflammatory and stromal cells, implying a co-inductive effect on tumor and stromal MMPs [6,7]. Furthermore, these localized increases in MMP activity can promote carcinogenesis by several mechanisms that include enabling tumor cell invasion of basement membranes and release of proangiogenic growth factors formerly sequestered in the ECM. Results from clinical studies, which show a positive correlation between the extent of MMP activation and cancer aggressiveness, provide pathophysiological relevance to perturbations in MMP regulation [6-8].
Native MMPs, which are secreted as membrane-bound proteins or released as proenzymes, require activation for proteolytic function [1]. MMP latency is attributable to a complex formed between a cysteine residue in the propeptide domain and the Zn2+ ion at the enzyme's catalytic site [9]. Consistent with the participation of MMPs in numerous physiological processes, pro-MMPs can be activated by a variety of mechanisms that include proteinases such as plasmin; thiol-oxidizing agents, e.g., HgCl2 and N-ethylmaleimide; low pH; and heat treatment [1]. MMP activation occurs in a sequential fashion, which initially entails intramolecular, followed by intermolecular, reactions and culminates in release of the propeptide's cysteine sulfur “blockage” of the active site [1]. Recently, the contribution of reactive oxygen and nitrogen species (ROS and RNS) in the regulation of pro-MMP activation and MMP gene expression has been recognized [10-13]. Consequently, conditions characterized by generation of high levels of ROS and RNS, which can readily oxidize thiols, can also promote MMP activation [10]. Furthermore, both clinical and experimental evidence implies that a perturbation in the local redox balance, such as could be induced by reactive species stress, serves to activate MMPs and thereby facilitate cancer progression [7,14,15]. These data help clarify how sustained inflammation, with its ensuing nitrosative and oxidative stress, is a risk factor for many human cancers, including HNSCC [7,14-16].
Despite the obvious importance of the propeptide's cysteine thiol in preserving MMP latency, limited studies have evaluated the effects of reduced thiols on MMP activation and function [13,17]. Okamoto et al. determined that glutathione (GSH), but not N-acetylcysteine (NAC), complexed with MMP-9's auto-inhibitory domain [13]. Other investigations by Albini et al. demonstrated that introduction of NAC to conditioned medium obtained from cultured Kaposi sarcoma cells inhibited MMP-2 and MMP-9 gelatinase activity and that treatment of cultured Kaposi sarcoma cells with NAC significantly suppressed tumor cell invasion [17].
Because of the importance of MMP dysregulation in cancer development, identification and evaluation of MMP inhibitors as cancer chemotherapeutic agents are currently active areas of investigation [18-20]. Further, the use of MMP inhibitors in a cancer prevention (“chemoprevention”) protocol is appealing due to the importance of basement membrane invasion in transformation of premalignant lesions, e.g., progression of oral epithelial dysplasia to HNSCC. For many premalignant diseases, chemopreventive compounds will need to be used for the remainder of the patient's life. It is therefore crucial to identify effective, yet well-tolerated, chemopreventives while concurrently developing strategies to most effectively deliver these agents. This study investigated the effects of two reduced nonprotein thiols, i.e., GSH and NAC, on activation, function, and cellular–ECM interactions of the basement-membrane-degrading gelatinase MMP-9. Our results demonstrate that NAC and GSH inhibit organomercurial activation of pro-MMP-9 by a mechanism that involves protein S-thiolation. Isoform specificity was apparent from the results of the gelatinase activity assays. Although GSH and NAC reduced MMP-2 function, significant inhibition by either NAC or GSH was noted only in the MMP-9 assays. Immunoblot analyses, which demonstrated that GSH binds at or near the enzyme's active-site Zn2+ molecule, provided mechanistic insights into how reduced thiols inhibit MMP-9 function. Cell invasion assays revealed that NAC enhanced endostatin's ability to inhibit HNSCC cell invasion of a simulated basement membrane [21,22]. Finally, the feasibility of NAC's docking at MMP-9's active-site Zn molecule was demonstrated by computational molecular modeling. Collectively, these data show that reduced nonprotein thiols suppress MMP-9 activation and function in both recombinant enzymes and human cancer cells and introduce the prospect for use of these agents as chemopreventive compounds.
Materials and methods
Oral squamous cell carcinoma cell lines
Five cell lines derived from HNSCCs of the tongue that developed in men between the ages of 25 and 70 years were obtained from the American Type Culture Collection. All of the SCC cell lines are aneuploid, are immortalized, have an epithelial morphology, and show growth rates ranging between 0.8 and 1.0 population doublings/day. Our laboratory has confirmed that these SCC cell lines retain many characteristics of oral mucosa, inclusive of preservation of antioxidant, cytoprotective enzymes and carcinogen-metabolizing capacities [23]. These cell lines were cultured in their optimal medium (DMEM/F12, 90%; heat-inactivated fetal bovine serum 10%) at 37°C, 5% CO2.
In order to determine whether GSH's and NAC's MMP modulatory effects extended to mesenchymal origin cancers, selected experiments used cell strains obtained from persons with AIDS-related Kaposi sarcoma (AIDS-KS). These AIDS-KS cell strains retain many properties manifest in the AIDS-KS tumor, inclusive of generation of high levels of reactive oxygen and nitrogen species [24]. AIDS-KS cells were cultured in their optimal supplemented M199 (GIBCO, Grand Island, NY, USA) medium, as previously described [24].
Rationale for the use of MMP-9
As both MMP-2 and MMP-9 facilitate cancer cell invasion of basement membranes, initial studies entailed concurrent experiments using both gelatinases. Whereas preliminary zymography studies revealed that GSH and NAC inhibited activation of both pro-MMP-2 and pro-MMP-9, the MMP activity assays showed that reduced thiols elicited appreciably greater effects on MMP-9 relative to MMP-2. Subsequent experiments therefore focused on thiol–MMP-9 interactions.
Determination of the effects of reduced thiols on activation of pro-MMPs
The effects of reduced nonprotein thiols on activation of human recombinant pro-MMP-9 (Calbiochem, La Jolla, CA, USA) were determined by gelatin zymography. MMP activation was achieved through introduction of the organomercurial compound p-aminophyenylmercuric acetate (APMA). The experimental groups for these assays were: (1) pro-MMP-9, (2) pro-MMP-9 + 1 mM APMA, (3) pro-MMP-9 + APMA + equimolar GSH, (4) pro-MMP-9 + APMA + equimolar NAC, (5) pro-MMP-9 + APMA + equimolar GSSG, (6) pro-MMP-9 + APMA + 2.5 mM GSH (25-fold excess), (7) pro-MMP-9 + APMA + 2.5 mM NAC, and (8) pro-MMP-9 + APMA + 2.5 mM GSSG. Pro-MMP-9 was activated by a 3-h incubation (37°C, 5% CO2) with APMA (1 mM final concentration), with introduction of NAC, GSH, or GSSG in the appropriate experimental groups. Samples were diluted in the sample buffer [50 mmol/L Tris (pH 6.8), 0.5% SDS, 10% glycerol, and 0.2% bromphenol blue] and separated by electrophoresis on a 7.5% polyacrylamide gel containing 1 mg/ml gelatin as substrate. Gels were washed in 2.5% Triton X-100 for 1 h at room temperature, then incubated overnight in a reaction buffer [50 nmol/L Tris (pH 7.6), 150 mmol/L NaCl, 2.5 mmol/L CaCl2, and 0.02% sodium azide], and stained with Coomassie blue solution (0.25% Coomassie Blue R250, 50% methanol, and 7.5% acetic acid). GSH, NAC, and GSSG were all obtained from Sigma Chemical Co. (St. Louis, MO, USA). Notably, the GSSG (No. G 6654) is specifically processed to remove any residual GSH.
Additional experiments were conducted on cultured HNSCC and AIDS-KS cells to determine NAC's effect in vivo on cellular production, release, and activation potential of gelatinases. HNSCC and AIDS-KS cells were cultured in a standard amount of complete medium, with the inclusion of 25 mM NAC in the treated cultures. After 24 h, the conditioned medium was collected and gelatinase activity determined by gelatin zymography.
Characterization of the interaction between GSH, NAC, and pro-MMP-9
The gelatin zymography results suggested that S-thiolation with the propeptide cysteine residue was the mechanism by which reduced thiols inhibited APMA enzyme activation. Experiments were therefore conducted using the commercially available biotinylated glutathiolation detection agent GSH ethyl ester, biotin amide (BioGee; Molecular Probes, Eugene, OR, USA), in conjunction with the thiol-reducing agent dithiothreitol (DTT) to determine whether GSH/NAC pro-MMP-9 interactions resulted in disulfide bond formation. Pro-MMP-9 was incubated (37°C, 3 h) with 100 μM BioGee, which in select groups was followed by addition of 2, 3, 5, or 10 μM DTT, with an additional 30-min incubation at 37°C. Samples were run on 7.5% native gels, with BioGee–MMP interactions identified by staining with LightShift-stabilized streptavidin–horseradish peroxidase (Pierce, Rockford, IL, USA). The membrane was then washed and probed with anti-MMP-9 antibody (Calbiochem, No. IM72), followed by an anti-mouse IgG horseradish peroxidase secondary antibody (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Immunoreactive signals were visualized by exposure to radiographic film.
Evaluation of the effects of reduced nonprotein thiols on MMP functional activities
The EnzChek Gelatinase/Collagenase Assay (Molecular Probes) was used to determine the effects of reduced thiols on functional activities of active human recombinant MMPs (Calbiochem). Due to the findings of Nyberg et al., which demonstrated endostatin–MMP interactions, endostatin was included in selected experimental groups [25]. The experimental groups consisted of: (1) MMP-9, (2) MMP-9 + equimolar GSH, (3) MMP-9 + equimolar NAC, (4) MMP-9 + equimolar GSSG, (5) MMP-9 + 100-fold excess GSH, (6) MMP-9 + 100-fold excess NAC, (7) MMP-9 + 100-fold excess GSSG, (8) MMP-9 + 250-fold excess GSH, (9) MMP-9 + 250-fold excess NAC, (10) MMP-9 + 250-fold excess GSSG, (11) MMP-9 + 500-fold excess GSH, (12) MMP-9 + 500-fold excess NAC, (13) MMP-9 + 500-fold excess GSSG, (14) MMP-9 + 10 μg/ml endostatin, (15) MMP-9 + 10 μg/ml endostatin + 250 × NAC, and (16) MMP-9 + 10 mM EDTA (negative control). A concurrently conducted collagenase standard curve (ranging from 0.00125 to 0.03 U) (1 U of activity was defined as the amount of enzyme required to liberate 1 μmol of L-leucine equivalents from collagen in 5 h at 37°C, pH 7.5) was conducted with each assay. After a 48-h incubation (37°C, 5% CO2) of enzyme with gelatinase substrate, fluorescence intensity (indicative of liberation of fluorescent tag due to proteolytic activity) was recorded and results were expressed as units of activity.
Assessment of the interactions between GSH, Zn2+, and MMP-9
Immunoblot analyses were conducted using the commercially available glutathiolation detection agent BioGee. The Zn chelator N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediame (TPEN) was included in some of the experimental groups in order to assess whether GSH–MMP-9 interactions entailed Zn interactions. The groups evaluated for these studies were: (1) active MMP-9, (2) active MMP-9 + 464 μM BioGee (800×), (3) active MMP-9 + 464 μM TPEN (800×) followed by the addition of BioGee (464 μM), and (4) MMP-9 + 464 μM BioGee followed by 464 μM TPEN. Similar experiments were also conducted using 1160 μM (2000×) TPEN or BioGee. Samples underwent a 2-h incubation (37°C) with or without TPEN or BioGee, followed by the addition of either BioGee or TPEN to the appropriate group and a second 2-h incubation. Samples were run on 7.5% nondenatured gels. The membrane was then washed and probed with an anti-MMP-9 antibody (Calbiochem). Light-Shift-stabilized streptavidin–horseradish peroxidase (Pierce Biotechnology) and anti-mouse IgG conjugated to horseradish peroxidase (Amersham) were used to detect GSH and MMP-9, respectively.
Determination of the effects of NAC and endostatin on cellular invasion
A series of in vivo experiments, which entailed cell invasion assays (No. QIA129; Calbiochem), was conducted to evaluate NAC's effect on HNSCC invasive capacity. NAC was used for these studies because NAC, unlike GSH, freely enters cells. Endostatin (Calbiochem) was included in some experimental groups, as results from our lab [21,22] and others [25] have shown that endostatin inhibits human cancer cell invasion. The experimental groups consisted of: (1) 48-h serum-deprived control, (2) 48-h serum deprived + 25 mM NAC during cell invasion, (3) 24-h serum deprived + 25 mM NAC during final 24 h serum deprivation and also during cell invasion, (4) 48 h serum deprivation, 30 min pretreatment with endostatin (10 μg/ml) with endostatin remaining present during invasion, (5) identical to 4 with the inclusion of 25 mM NAC during cell invasion, and (6) 48 h serum deprivation with final 24 h containing 25 mM NAC, 30 min endostatin pretreatment, and fresh 25 mM NAC added during the invasion assay. Invasion chambers containing cells (3.5 × 105) were incubated for 48 h under standard conditions (5% CO2, 37°C), with the chemoattractant (DMEM/F12 + 10% heat-inactivated fetal bovine serum) present in the lower chamber. As we have previously observed a cell-strain-associated difference in calcein loading [21,22], a same-cell-line standard curve was conducted with each assay. Results were expressed as numbers of successfully invaded cells as determined by sample fluorescence intensity relative to its matched standard curve.
Computational molecular modeling to assess feasibility of NAC docking to active-site Zn molecule
The NAC molecule was computationally docked with the MMP-9 protein using MacroModel 8.0 (Schrodinger, LLC). A conformational search was performed using the Monte Carlo molecular mechanics minimization method [26] with the OPLS-AA force field [27]. The NAC molecule was fully flexible and the MMP-9 protein frozen throughout the conformational search. The number of conformational search steps was set at 1000. The MMP-9 monomer structure was taken from the Protein Data Bank structure Accession No. 1GKC [28] edited to remove all water molecules and the bound inhibitor. The initial structure for NAC was built using GaussView (Gaussian, Inc.) and the geometry was optimized using density functional theory at the B3LYP/6-31 + G* level of theory with the Gaussian03 software package [29]. All computations were performed on a Silicon Graphics, Inc., Octane2 computer running the IRIX operating system.
Statistical analyses
The Yates corrected χ2 test was used to assess the effects of reduced nonprotein thiols on MMP-9 enzyme function and invasion of HNSCC tumor cells. The level of significance was established at the 95% confidence interval (α<0.05).
Results
Reduced, nonprotein thiols inhibit APMA-induced activation of pro-MMP-9
Gelatin zymography results demonstrated that inclusion of either 2.5 mM GSH or 2.5 mM NAC inhibited APMA-induced activation of pro-MMP-9 (Fig. 1A). In contrast, addition of 2.5 mM GSSG did not affect APMA-induced activation of MMP-9 (Fig. 1A). Equimolar levels of either GSH or NAC did not affect APMA-mediated MMP-9 activation. Further, although GSH and NAC did reduce APMA activation of pro-MMP-2, the extent of inhibition was appreciably less than the inhibitory effects noted with MMP-9 (data not shown).
Fig. 1.

Reduced nonprotein thiols inhibit activation of pro-MMP-9 by a mechanism that entails S-thiolation. (A) The effects of nonprotein thiols on APMA-mediated activation of pro-MMP-9 were determined by gelatin zymography. The enzyme-digested bands are observed as white bands against a dark background. Lane assignments: (1) pro-MMP-9, (2) pro-MMP-9 + 2.5 mM GSH, (3) pro-MMP-9 + 2.5 mM NAC, (4) pro-MMP-9 + 2.5 mM GSSG, (5) pro-MMP-9 + APMA, (6) pro-MMP-9 + 2.5 mM GSH + APMA, (7) proMMP-9 + 2.5 mM NAC + APMA, (8) pro-MMP-9 + 2.5 mM GSSG + APMA. (B) The nature of the reduced thiol pro-MMP-9 interaction was assessed by inclusion of the biotinylated GSH analogue, BioGee, followed by introduction of the disulfide-reducing agent, DTT (blot a). Western blot analyses were then conducted on the same washed membrane to confirm the integrity of the proMMP-9 protein (blot b). Lane assignments: (1) pro-MMP-9 only, (2) pro-MMP-9 + 100 μM BioGee, (3) pro-MMP-9 + 100 μM BioGee + 2 mM DTT, (4) proMMP-9 + 100 μM BioGee + 3 mM DTT, (5) pro-MMP-9 + 100 μM BioGee + 5 mM DTT, (6) pro-MMP-9 + 100 μM BioGee + 10 mM DTT. (C and D) Gelatin zymography was used to assess the effects of reduced thiols on release and subsequent activation of MMP-2 and MMP-9 which was secreted into the conditioned medium by cultured HNSCC cells as well as (E) cells isolated from AIDS-related Kaposi sarcoma (AIDS-KS). To enhance cellular release of MMPs, HNSCC and AIDS-KS cultures were serum deprived (SD) for 48 h, and conditioned medium was collected during this time. Lane assignments: (C) (1) HNSCC 4 48 h SD, (2) HNSCC 4 24 h SD + 24 h 25 mM NAC, (3) HNSCC 4 48 h SD + APMA, (4) HNSCC 4 24 h SD + 24 h 25 mM NAC + APMA, (5) MMP-2/MMP-9 standards; (D) (1) HSCC 9 24 h SD, (2) HNSCC 9 24 h SD + 24 h + 25 mM NAC, (3) HNSCC 9 48 h SD + APMA, (4) HNSCC 9 24 h SD + 24 h 25 mM NAC + APMA, (5) MMP-2/MMP-9 standards; (E) (1) KS 48 h SD, (2) KS 24 h SD + 25 mM NAC, (3) KS 48 h SD + APMA, (4) KS 24 h SD + 24 h 25 mM NAC + APMA, (5) MMP-2/MMP-9 standards.
Reduced thiol–pro-MMP-9 interactions involve disulfide bond formation
The introduction of DTT after BioGee–pro-MMP-9 interaction resulted in a dose-dependent decrease in binding of biotinylated GSH (Fig. 1B, blot a) while preserving the integrity of the pro-MMP-9 protein (Fig. 1B, blot b). These data are consistent with DTT-mediated reduction of a disulfide bond generated as a consequence of S-thiolation.
NAC inhibits production and subsequent activation of pro-MMP-2 and pro-MMP-9 in cultured HNSCC and AIDS-KS cells
Inclusion of NAC with cultured HNSCC cells affected gelatinase activities in a bimodal fashion, i.e., a reduction in the amount of pro-MMP-2 and pro-MMP-9 secreted into the conditioned medium and the presence of NAC in the conditioned medium also inhibited APMA-mediated activation of both gelatinases (Figs. 1C and 1D). Similar suppressive effects were observed in all five HNSCC cell lines. MMP-2 was the primary gelatinase released from AIDS-KS cells. Similar to the HNSCC findings, the AIDS-KS results revealed that NAC treatment reduced both the amount of MMP protein released and its responsiveness to APMA-induced activation (Fig. 1E). Comparable inhibitory effects were observed after NAC treatment of three different AIDS-KS cell strains. Finally, inclusion of NAC did not result in an overall reduction in cell protein, as total protein levels were comparable among the NAC-treated and control cultures.
GSH and NAC also significantly inhibit active MMP-9 function
Results from the MMP-9 functional activity assays revealed several, compound-associated findings (Table 1). Inclusion of either GSH or NAC, at concentrations of 100× and greater, resulted in significant inhibition of MMP-9 function. There were, however, differences in GSH- and NAC-associated effects. Relative to GSH, NAC's inhibitory effect was higher at equimolar concentrations, and its effectiveness peaked at 250× levels. In contrast, GSH inclusion demonstrated a dose-dependent effect, with the greatest inhibition detected at the highest (500×) GSH level. Inclusion of GSSG was associated with some statistically insignificant inhibition of MMP-9 function. Inclusion of endostatin alone had no effect on MMP-9 function, whereas the effects of the NAC (250×) + endostatin combination were slightly lower but comparable to NAC treatment (Table 1).
Table 1.
Effects of GSH, NAC, GSSG, and endostatin on MMP-9 functional activity
| Experimental group |
Equimolar | 100× | 250× | 500× |
|---|---|---|---|---|
| GSH | −7.1 ± 10.3 | −47.3 ± 5.6 * | −52.8 ± 6.8 * | −60.6 ± 4.7 * |
| NAC | −22.0 ± 9.8 | −47.3 ± 4.3 * | −61.5 ± 11.6 * | −57.2 ± 6.3 * |
| GSSG | −8.3 ± 9.4 | −12.9 ± 10.6 | −13.0 ± 11.0 | −1.1 ± 13.2 |
| Single-dose experiments | ||||
| Endostatin (10 μg/ml) | +4.2 ± 14.4 | |||
| Endostatin (10 μg/ml) + 250 × NAC | −43.5 ± 9.0 | |||
| EDTA (10 mM) | −95.4 ± 0.02 | |||
N = 5 for each experimental group, mean percentage change in MMP-9 activity ± SEM.
p < 0.05, Yates corrected χ2 test.
GSH–MMP-9 interactions entail Zn interactions
Co-incubation of active MMP-9 with the biotinylated GSH analogue, BioGee, resulted in formation of a complex that confirmed GSH-MMP-9 binding (Figs. 2A and 2B). As would be anticipated, MMP-9–GSH binding intensity increased with the higher BioGee concentration. Incubation of MMP-9 with the zinc chelator TPEN before introduction of BioGee appreciably inhibited GSH–MMP-9 complex formation. Furthermore, TPEN addition after BioGee introduction also reduced GSH–MMP-9 complex formation, albeit to a lesser extent than TPEN introduction before BioGee (Fig. 2A).
Fig. 2.

Reduced nonprotein thiols interact with MMP-9′s active-site Zn2+ molecule. (A) Immunoblot analyses were conducted using the biotinylated GSH analogue, BioGee, to determine the interaction of reduced nonprotein thiols with the active-site Zn molecule. (B) Western blot analyses were then conducted on the same washed membrane to confirm the integrity of the MMP-9 protein. Lane assignments were: (1) active MMP-9 with no incubation, (2) active MMP-9 + vehicle (ethanol) control with no incubation, (3) active MMP-9 + vehicle + 4 h incubation, (4) active MMP-9 + 464 μM (800×) BioGee, (5) active MMP-9 + 464 μM TPEN first, followed by 464 μM BioGee, (6) active MMP-9 + 464 μM BioGee first, followed by 464 μM TPEN, (7) active MMP-9 + 1160 μM (2000×) BioGee, (8) active MMP-9 + 1160 μM TPEN first, followed by 1160 μM BioGee, and (9) active MMP-9 + 1160 μM BioGee first, followed by 1160 μM TPEN.
The actions of endostatin and NAC are additive in their ability to inhibit invasion of human HNSCCs
Consistent with our previous findings, there were appreciable (fivefold) cell-line-associated differences in cellular invasive capacities (means ranged from 874 to 4539) [22]. Interestingly, the cell line (HNSCC 4) that showed the greatest cell invasive capacity was also the exclusive cell line in which all treatments suppressed cell invasion. Our results also show that the timing of agent introduction as well as agent combinations affected the experimental outcome (Fig. 3). The average cumulative effect of inclusion of NAC just before invasion and endostatin pretreatment alone was a slight, insignificant increase in invasive capacity (Fig. 3). Further, whereas NAC (pretreatment and during invasion) and the endostatin + NAC combination (30 min endostatin pretreatment and NAC only during invasion) both reduced cell invasion, the differences were not statistically significant. Notably, the combination of 25 mM NAC (24 h pretreatment, NAC also present during invasion) with a 30-min pretreatment with endostatin (10 μg/ ml) inhibited HNSCC invasive capacity in each cell line in every experiment (n=8, p<0.005, mean percentage decrease 53.0 ± 6.5%, SEM).
Fig. 3.

N-acetylcysteine augments endostatin's ability to suppress invasion of HNSCC cells. The ability of NAC and endostatin to inhibit the ability of calcein-loaded HNSCC cells to invade a synthetic basement membrane was evaluated using a commercially available cell invasion assay kit. Notably, in each cell line (n = 5) and in every experiment conducted (n = 8), the combination of NAC pretreatment with inclusion of NAC and endostatin during invasion inhibited HNSCC invasive capacities (p ≤ 0.05, Yates corrected χ2 test).
Computer modeling of NAC–MMP-9 interactions
Computer modeling results demonstrate that NAC is capable of undergoing a slow binding reaction with the catalytic-site Zn (Fig. 4). Additional computational molecular modeling studies depict how NAC is capable of docking at the MMP-9 active-site Zn molecule (Fig. 5).
Fig. 4.
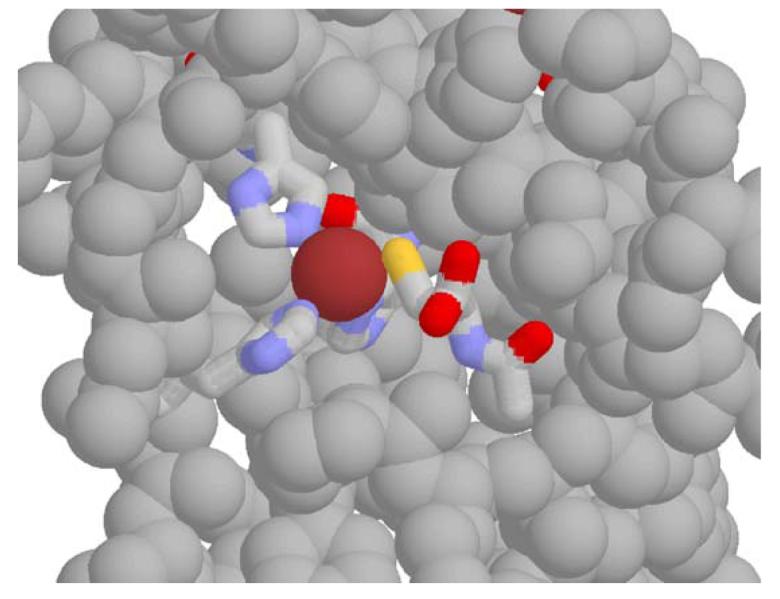
A model for the complex of active MMP-9 with N-acetylcysteine. The atomic coordinates used for the protein are from the structure of pro-MMP-9 (Accession No. 1L6J in the Protein Data Bank), with the prosequence 1–105 removed. The inhibitor is modeled into the groove on the surface of the protein that is exposed upon loss of the prosequence, in a position analogous to that of Cys 99, which in the structure is coordinated to the active-site zinc. N-acetylcysteine is proposed to coordinate in a similar fashion, as shown. The remaining ligands of the zinc are, clockwise from bottom left, His 411, His 405, and His 401 (positioned behind the zinc). We speculate that NAC (and also GSH) undergoes a slow binding reaction with the catalytic-site Zn2+, resulting in formation of a nonprotein thiol–Zn2+ complex.
Fig. 5.
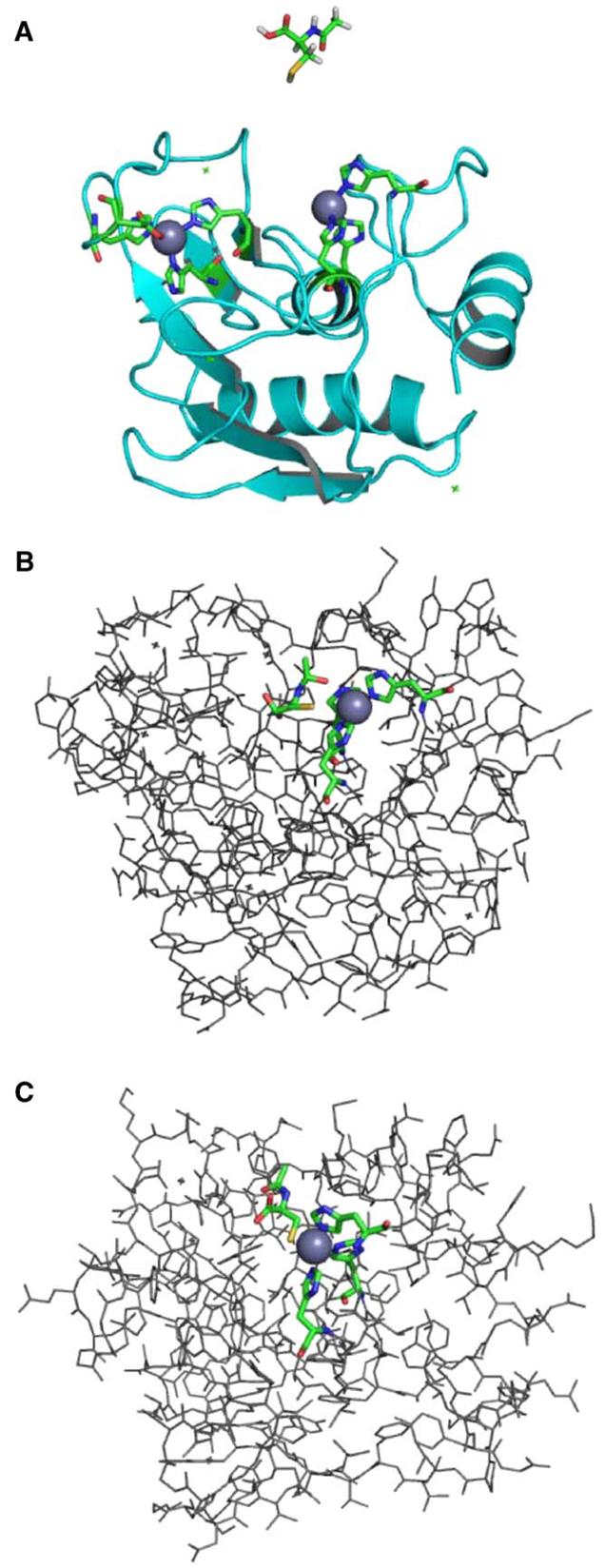
Docking of the MMP-9/NAC complex. (A) MMP-9 protein structure showing both zinc sites. The active-site zinc (on right, gray sphere), within the potential docking cleft, is coordinated by three histidines represented as sticks with the following atom coloring: carbon—green, nitrogen—blue, oxygen—red. Hydrogen atoms are not shown in the protein structure. The NAC molecule used in the docking study is shown above the MMP-9 structure. The NAC geometry was optimized at the B3LYP/6-31 + G* level of theory using Gaussian03 software [29]. The atom coloring is as above with the following additions: sulfur—yellow, hydrogen—white. (B) A docked structure of the MMP-9/NAC complex showing NAC positioned in the docking cleft nearest the active-site zinc, with the NAC sulfur within coordination distance of the metal. This structure was determined using the computational docking procedure described in the text. Hydrogen atoms are not shown. Atom coloring is as described above. (C) A model of the MMP-9/NAC complex in which the NAC ligand is coordinated to the active-site zinc analogous to Cys 99 in the crystal structure of pro-MMP-9 taken from the Protein Data Bank (Accession No. 1L6J) [45]. A partial geometry optimization was performed using the ONIOM method [46] as implemented in Gaussian03 [29] at the RHF/LANL2MB:UFF level of theory. The NAC ligand, zinc atom, and three coordinated histidines were included in the quantum mechanical region (RHF/LANL2MB), and the remaining portion of the MMP-9 protein was treated at the molecular mechanics level (UFF). Atom coloring is as described above. All graphical representations were generated with PyMol [47].
Discussion
The oxidative stress that ensues during sustained inflammation perturbs the local redox balance, thereby establishing conditions that favor MMP activation [10]. Notably, increased MMP activity can promote carcinogenesis by numerous mechanisms that include enabling migration and invasion of cancer cells, release of sequestered growth factors, and facilitating tumor-associated angiogenesis. This study investigated the effects of two reduced nonprotein thiols, i.e.; GSH and NAC, on the activation and function of the basement-membrane degrading gelatinase MMP-9.
Gelatin zymography results demonstrate that GSH and NAC, but not the oxidized GSSG, rendered pro-MMP-9 refractory to APMA-induced activation. These data implied that protein Sthiolation had occurred at the propeptide cysteine, preventing release of the thiol propeptide from the Zn atom and thereby preserving the S “blockage” of the enzyme's active site. Additional experiments, which demonstrated that the disulfide-reducing agent DTT reversed GSH pro-MMP binding, supported this premise. These data, which show that protein S-thiolation inhibits APMA-induced MMP activation, conflict with the findings of Okamoto et al., who observed MMP activation after S-glutathiolation [13]. There are, however, marked differences in the experimental designs. Okamoto et al. introduced GSH and peroxynitrite (ONOO−) concurrently and observed that the greatest MMP activation ensued after inclusion of equimolar levels of GSH and ONOO−. The results of Okamoto et al. suggest that the contribution of GSNO2 and subsequent S-oxide formation are attributable to their observed MMP activation [13]. Other differences, which emphasize the importance of experimental conditions, are apparent between Okamoto's and the current study. Whereas Okamoto et al. did not observe any effects after addition of NAC or any interactions between GSH and functional MMPs, our data show that both NAC and GSH are effective inhibitors of gelatinase activation and function, and our immunoblot results confirm that GSH interacts with active MMP-9. Concurrent experiments that were conducted on cultured HNSCC and AIDS-KS cells demonstrated in vivo efficacy as the introduction of NAC inhibited secretion and APMA activation of both pro-MMP-2 and pro-MMP-9. These data, which demonstrate NAC's effects on gene expression, likely reflect NAC's antioxidant properties and subsequent quenching of redox-mediated cell signaling. Notably, additional experiments to demonstrate S-thiolation of gelatinases released from cultured cells were unsuccessful due to: (1) an inability to obtain sufficient protein from conditioned medium with the use of separation beads and (2) protein destabilization during medium concentration.
The gelatinase functional assays demonstrated that the inhibitory effects of GSH and NAC are isoform specific, as these compounds significantly inhibited only MMP-9 activity. Although NAC and GSH did suppress MMP-2 activity, the extent of inhibition was not significant. Results from the immunoblot analyses showed that pretreatment with the Zn chelator TPEN reduced GSH–MMP-9 complex formation. These data imply that MMP-9's secondary and/or tertiary structure renders the active-site Zn accessible to reactions with nonprotein reduced thiols and that Zn–thiol binding restricts substrate–active site interactions. Shown in Fig. 4 is a molecular model that depicts how NAC could complex with the active site of MMP-9, whereas Fig. 5 demonstrates the feasibility of NAC docking at MMP-9's active-site Zn molecule. This premise is supported by the studies of Kleifeld et al., who clarified the nature of MMP-9 activation, i.e., the latent Zn binding site becomes catalytically competent by dissociation of the thiol propeptide from the Zn atom [30]. Although our data suggest a plausible MMP-9 inhibitory mechanism, in order to provide conclusive evidence for GSH's and NAC's interactions with MMP-9's active site, it is necessary to conduct quantitative biophysics. Not surprisingly, a common feature among many MMP inhibitors is the presence of a metal-binding group (often a carboxyl thiol, or hydroxamate) that chelates Zn [31]. Although previous studies by Nyberg et al. demonstrated that endostatin inhibited activation of pro-MMP-9 [25] we did not observe this effect, nor did we note any effect between endostatin and mature enzyme. These differences may reflect structural variations in the form of endostatin used, as its monomer is speculated to exert different effects relative to the NC1 trimer.
A large number of proteins are capable of undergoing protein S-thiolation. Demonstration of in vivo efficacy, however, is generally considered essential for this reaction to be considered consequential [32]. Results from our cell invasion studies show appreciable, cell-line-associated differences in invasive capacity and responsiveness to MMP inhibition. These variations likely reflect differences in cellular capacity to undergo transition to an invasive “amoeboid” phenotype (which permits cells to squeeze through matrix gaps when MMPs are blocked) [33] and also the specific cell line dependence on MMP-9 for basement membrane degradation. Furthermore, although our study used NAC levels comparable to those employed by Albini et al. [17], we did not observe significant inhibitory effects with NAC treatment only. These differences likely reflect variations in invasive properties of the cell populations tested. Albini et al. investigated NAC's effects on the invasion of Kaposi sarcoma (KS) cells [17]. KS cells are weakly tumorigenic and often require supplements, such as controlled release of angiogenic compounds, to induce KS tumors in nude mice [34]. In contrast, the HNSCC cell lines evaluated in the current study are highly tumorigenic, locally invasive, and capable of forming distant metastases in vivo [35]. In the current study, significant inhibition was obtained only during concurrent treatment with NAC and endostatin. This additive effect reflects the agents' different mechanisms of action, as endostatin inhibits cancer cell invasion by a mechanism independent of MMP inhibition, i.e., binding to tropomyosin myofilaments [21,22], and as shown by our gelatin zymography data, NAC inhibits HNSCC gelatinase release. Notably, these data, which show significant effects only with combination therapy, are consistent with results obtained from human clinical trials. Single-agent human clinical studies to assess the efficacy of inhibitors of MMPs and serine proteinases, as well as endostatin, have not been promising [36-39]. Collectively, these findings imply that combinations of agents in conjunction with innovative drug delivery methods have the greatest promise for clinical success in cancer therapy.
MMP activation occurs after limited proteolysis of zymogens or by chemical modifications induced by agents such as APMA or reactive species [1]. A uniform feature of these activating agents is oxidation of the inhibitory peptide cysteine. Due to the relatively sequestered position of the propeptide cysteine in the native pro-MMP-9 protein, access for NAC or GSH would likely be restricted. A more probable scenario is that GSH and NAC react with the propeptide cysteine after partial activation has rendered the propeptide cysteine accessible to S-thiolation, which stabilizes the inhibitory peptide cysteine and prevents further oxidation and formation of the potential MMP-activating sulfenic, sulfinic, and sulfonic acids [32]. Notably, of the 19 cysteine residues present in MMP-9, only the cysteine residue in the propeptide domain that coordinates with Zn in the active site has been shown to be irreversibly oxidized to a sulfinic or sulfonic residue [12]. Furthermore, the results of Okamoto et al. suggest that high, redox-scavenging, local levels of reduced nonprotein thiols can prevent the irreversible, MMP-activating oxidations induced by reactive nitrogen species [13].
Whereas TIMPs can inhibit MMP-9 in an ∼1:1 molar ratio, the levels of reduced thiols used to induce MMP inhibitory effects in this study were appreciably higher. Locally injectable controlled release delivery vehicles, however, can provide therapeutically relevant agent concentrations at the treatment site without inducing deleterious systemic effects such as impaired wound healing [40,41]. Controlled local release of reduced nonprotein thiols could inhibit MMP activity by several mechanisms, i.e., by altering the redox balance, which would quench intracellular signaling and decrease MMP activation, and by direct inhibition of gelatinase activation and function. In addition, high tissue levels of reduced thiols have the potential to directly remove the catalytic Zn from its MMP active site. Although GSH reacts with a wide variety of redox-sensitive metal ions, the slow dissociation rate, reflective of the stability of the GSH–Zn2+ complex, makes zinc interactions unique [42].
Both tolerance and efficacy, however, are important considerations when evaluating potential chemopreventive compounds. Notably, GSH and NAC are naturally occurring compounds, and administration of NAC to humans for management of pulmonary diseases and nephrotoxicity has been effectively used and well tolerated for years [43,44]. In conclusion, these data suggest that by virtue of their redox-modulating and gelatinase-inhibitory properties, GSH and NAC represent promising compounds which merit consideration for controlled-release, site-specific, chemopreventive applications.
Acknowledgments
These studies were supported by NIH RO1 CA95901 and R21 CA111210 (S. R. Mallery).
Abbreviations
- ECM
extracellular matrix
- MMP
matrix metalloproteinase
- GSH
glutathione
- GSSG
glutathione disulfide
- NAC
N-acetylcysteine
- APMA
p-aminophyenylmercuric acetate
- DTT
dithiothreitol
- TPEN
N,N′, N′-tetrakis(2-pyridylmethyl)ethylenediame
- HNSCC
head and neck squamous cell carcinoma
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- TIMP
tissue inhibitor of matrix metalloproteinases
References
- 1.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function and biochemistry. Circ. Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 2.Westermark J, Kahari VM. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 1999;13:781–792. [PubMed] [Google Scholar]
- 3.Blavier L, Henriet P, Imren S, Declerck YA. Tissue inhibitors of matrix metalloproteinases in cancer. Ann. N.Y. Acad. Sci. 1999;878:108–119. doi: 10.1111/j.1749-6632.1999.tb07677.x. [DOI] [PubMed] [Google Scholar]
- 4.Curran S, Marray GI. Matrix metalloproteinases in tumour invasion and metastasis. J. Pathol. 1999;189:300–308. doi: 10.1002/(SICI)1096-9896(199911)189:3<300::AID-PATH456>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 5.Garbett EA, Reed MW, Brown NJ. Proteolysis in human breast and colorectal cancer. Br. J. Cancer. 1999;81:287–293. doi: 10.1038/sj.bjc.6690689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kurahara S, Shinohara M, Ikebe T, Nakamura S, Beppu M, Hiraki A, Takeuchi H, Shirasuna K. Expression of MMPs, MT-MMP, and TIMPs in squamous cell carcinoma of the oral cavity: correlations with tumor invasion and metastasis. Head Neck. 1999;21:617–638. doi: 10.1002/(sici)1097-0347(199910)21:7<627::aid-hed7>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 7.Franshi A, Santucci M, Masini E, Sardi I, Paglierani M, Gallo O. Expression of matrix metalloproteinase 1, matrix metalloproteinase 2, and matrix metalloproteinase 9 in carcinoma of the head and neck. Cancer. 2002;95:1902–1910. doi: 10.1002/cncr.10916. [DOI] [PubMed] [Google Scholar]
- 8.Jordan RC, Macebeo-Ong M, Shiboski CH, Dekker N, Ginzinger DG, Wong DTW, Schmidt BL. Overexpression of matrix metalloproteinase-1 and -9 mRNA is associated with progression of oral dysplasia to cancer. Clin. Cancer Res. 2004;10:6460–6465. doi: 10.1158/1078-0432.CCR-04-0656. [DOI] [PubMed] [Google Scholar]
- 9.Van Wart H, Birkedal-Hansen H. The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA. 1990;87:5578–5582. doi: 10.1073/pnas.87.14.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson KK, Melendez A. Mitochondrial redox control of matrix metalloproteinases. Free Radic. Biol. Med. 2004;37:768–784. doi: 10.1016/j.freeradbiomed.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Ishii Y, Ogura T, Tatemichi M, Jufisawa H, Otsuka F, Esumi H. Induction of matrix metalloproteinase gene transcription by nitric oxide and mechanisms of MMP-1 gene induction in human melanoma cell lines. Int. J. Cancer. 2003;103:161–168. doi: 10.1002/ijc.10808. [DOI] [PubMed] [Google Scholar]
- 12.Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton AS. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- 13.Okamoto T, Akaike T, Sawa T, Miyamoto Y, van der Vliet A, Maeda H. Activation of matrix metalloproteinases by peroxynitrite-induced protein S-glutathionylation via disulfide S-oxide formation. J. Biol. Chem. 2001;276:29596–29602. doi: 10.1074/jbc.M102417200. [DOI] [PubMed] [Google Scholar]
- 14.Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis. 2000;21:361–370. doi: 10.1093/carcin/21.3.361. [DOI] [PubMed] [Google Scholar]
- 15.Wink DA, Vodovotz Y, Laval J, Laval F, Dewhirst MW, Mitchell JB. The multifaceted roles of nitric oxide in cancer. Carcinogenesis. 1998;19:711–721. doi: 10.1093/carcin/19.5.711. (Franchi et al.; Cancer 2002) [DOI] [PubMed] [Google Scholar]
- 16.McCord JM, Edeas MA. SOD, oxidative stress and human pathologies: a brief history and a future direction. Biomed. Pharmacother. 2005;59:139–142. doi: 10.1016/j.biopha.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Albini A, Morini M, D'Agostini F, Ferrari N, Campelli F, Arena G, Noonan DM, Pesce C, De Flora S. Inhibition of angiogenesis-driven Kaposi's sarcoma tumor growth in nude mice by oral N-acetylcysteine. Cancer Res. 2001;61:8171–8178. [PubMed] [Google Scholar]
- 18.Cianfrocca M, Cooley TP, Lee JY, Rudek MA, Scadden DT, Ratner L, Pluda JM, Figg WD, Krown SE, Dezube BJ. Matrix metalloproteinase inhibitor COL-3 in the treatment of AIDS-related Kaposi's sarcoma: a phase I AIDS malignancy consortium study. J. Clin. Oncol. 2002;20:153–159. doi: 10.1200/JCO.2002.20.1.153. [DOI] [PubMed] [Google Scholar]
- 19.Maquoi E, Sounni NE, Devy L, Olivier F, Frankenne F, Krell HW, Grams F, Foidart JM, Noel A. Anti-invasive, antitumoral, and antiangiogenic efficacy of a pyrimidine-2,3,6-trione derivative, an orally active and selective matrix metalloproteinase inhibitor. Clin. Cancer Res. 2004;10:4038–4047. doi: 10.1158/1078-0432.CCR-04-0125. [DOI] [PubMed] [Google Scholar]
- 20.Ikejiri M, Bernardo MM, Bonfil RD, Toth M, Chang M, Fridman R, Mobashery S. Potent mechanism-based inhibitors for matrix metalloproteinases. J. Biol. Chem. 2005;280:33992–34002. doi: 10.1074/jbc.M504303200. [DOI] [PubMed] [Google Scholar]
- 21.Mallery SR, Morse MA, Wilson RF, Pei P, Ness GM, Bradburn JE, Renner RJ, Schuller DE, Robertson FM. AIDS-related Kaposi's sarcoma cells rapidly internalize endostatin, which co-localizes to tropomyosin microfilaments and inhibits cytokine-mediated migration and invasion. J. Cell. Biochem. 2003;89:133–143. doi: 10.1002/jcb.10489. [DOI] [PubMed] [Google Scholar]
- 22.Wilson RF, Morse MA, Pei P, Renner RJ, Schuller DE, Robertson FM, Mallery SR. Endostatin inhibits migration and invasion of head and neck cancer cells. Anticancer Res. 2003;23:1289–1296. [PubMed] [Google Scholar]
- 23.Rinaldi AL, Morse MA, Fields HW, Rothas DA, Pei P, Rodrigo KA, Renner RJ, Mallery SR. Curcumin activates the aryl hydrocarbon receptor yet significantly inhibits (−)-benzo(a)pyrene-7Rtrans-7,8-dihydrodiol bioactivation in oral squamous cell carcinoma cells and oral mucosa. Cancer Res. 2002;62:5451–5456. [PubMed] [Google Scholar]
- 24.Mallery SR, Pei P, Landwehr DJ, Clark CM, Bradburn JE, Ness GM, Robertson FM. Implications for oxidative and nitrosative stress in the pathogenesis of AIDS-related Kaposi's sarcoma. Carcinogenesis. 2004;25:597–603. doi: 10.1093/carcin/bgh042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nyberg P, Heikkila P, Sorsa TM, Luostarinen J, Heljasvaara R, Stenman U-H, Pihlajaniemi T, Salo T. Endostatin inhibits human tongue carcinoma cell invasion and intravasation and blocks the activation of matrix metalloprotease-2, -9, and -13. J. Biol. Chem. 2003;278:22404–22411. doi: 10.1074/jbc.M210325200. [DOI] [PubMed] [Google Scholar]
- 26.Chang G, Guida WC, Still WC. An internal coordinate Monte-Carlo method for searching conformational space. J. Am. Chem. Soc. 1989;111:4379–4386. [Google Scholar]
- 27.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11235. [Google Scholar]
- 28.Rowsell S, Hawtin P, Minshull CA, Jepson H, Brockbank SMV, Barrat DG, Slater AM, McPheat WL, Waterson D, Henney AM, Pauptit RA. Crystal structure of human MMP9 in complex with a reverse hydroxamate inhibitor. J. Mol. Biol. 2002;319:173–181. doi: 10.1016/S0022-2836(02)00262-0. [DOI] [PubMed] [Google Scholar]
- 29.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr., Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, revision C.02. Gaussian; Wallingford, CT: 2004. [Google Scholar]
- 30.Kleifeld O, Van Den Steen PE, Frenkel A, Chang F, Jiang HL, Opdenakker G, Sagi I. Structural characterization of the catalytic active site in the latent and active natural gelatinase B from human neutrophils. J. Biol. Chem. 2000;275:34335–34343. doi: 10.1074/jbc.M005714200. [DOI] [PubMed] [Google Scholar]
- 31.Liekens S, De Clercq E, Neyts J. Angiogenesis: regulators and clinical applications. Biochem. Pharmacol. 2001;61:253–270. doi: 10.1016/s0006-2952(00)00529-3. [DOI] [PubMed] [Google Scholar]
- 32.Shackelford RE, Heinloth AN, Heard S, Paules RS. Cellular and molecular targets of protein S-glutathiolation. Antioxid. Redox Signaling. 2005;7:940–950. doi: 10.1089/ars.2005.7.940. [DOI] [PubMed] [Google Scholar]
- 33.Wolf K, Mazo I, Leung H, Engelke K, von Andrian U, Deryugina E, Strongin A, Brocker E-B, Friedl P. Compensation mechanism in tumor cell migration: mesenchymal–amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 2003;160:267–277. doi: 10.1083/jcb.200209006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mallery SR, Pei P, Kang J, Zhu G, Ness GM, Schwendeman SP. Sustained angiogenesis enables in vivo transplantation of mucocutaneous derived AIDS-related Kaposi's sarcoma cells in murine hosts. Carcinogenesis. 2000;21:1647–1653. doi: 10.1093/carcin/21.9.1647. [DOI] [PubMed] [Google Scholar]
- 35.Mallery SR, Shenderova A, Pei P, Begum S, Ciminieri JR, Wilson RF, Casto BC, Schuller DE, Morse MA. Effects of 10-hydroxycamptothecin, delivered from locally injectable poly(lactidecoglycolide) microspheres, in a murine human oral squamous cell carcinoma regression model. Anticancer Res. 2001;21:1713–1722. [PubMed] [Google Scholar]
- 36.Mannello F, Tonti G, Papa S. Matrix metalloproteinase inhibitors as anticancer therapeutics. Curr. Cancer Drug Targets. 2005;5:285–298. doi: 10.2174/1568009054064615. [DOI] [PubMed] [Google Scholar]
- 37.Molina JR, Reid JM, Erlichman C, Sloan JA, Furth A, Safgren SL, Lathia CD, Alberts SR. A phase I and pharmacokinetic study of the selective, non-peptide inhibitor of matrix metalloproteinase BAY 12-9566 I combination with etoposide and carboplatin. Anticancer Drugs. 2005;16:997–1002. doi: 10.1097/01.cad.0000176504.86551.5c. [DOI] [PubMed] [Google Scholar]
- 38.Sridhar SS, Shaepherd FA. Targeting angiogenesis: a review of angiogenesis inhibitors in the treatment of lung cancer. Lung Cancer. 2003;42(Suppl 1):S81–S91. doi: 10.1016/s0169-5002(03)00308-8. [DOI] [PubMed] [Google Scholar]
- 39.Thomas JP, Arzoomanian RZ, Alberti D, Marnocha R, Lee F, Friedl A, Tutsch K, Dresen A, Geiger P, Pluda J, Fogler W, Schiller JH, Wilding G. Phase I pharmacokinetic and pharmacodynamic study of recombinant human endostatin in patients with advanced solid tumors. J. Clin. Oncol. 2003;21:223–231. doi: 10.1200/JCO.2003.12.120. [DOI] [PubMed] [Google Scholar]
- 40.Varde NK, Pack DW. Microspheres for controlled release drug delivery. Expert Opin. Biol. Ther. 2004;4:35–51. doi: 10.1517/14712598.4.1.35. [DOI] [PubMed] [Google Scholar]
- 41.Schwendeman SP. Recent advances in the stabilization of proteins encapsulated in injectable PLGS delivery systems. Crit. Rev. Ther. Drug Carrier Syst. 2002;19:73–98. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.20. [DOI] [PubMed] [Google Scholar]
- 42.Mladenov M, Mirceski V, Gjorgoski I, Jordanoski B. Bioelectrochemistry. 2004;65:69–76. doi: 10.1016/j.bioelechem.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 43.Tepel M, Zidek W. N-acetylcysteine in nephrology; contrast nephrology and beyond. Curr. Opin. Nephrol. Hypertens. 2004;13:649–654. doi: 10.1097/00041552-200411000-00011. [DOI] [PubMed] [Google Scholar]
- 44.Kelly GS. Clinical applications of N-acetylcysteine. Altern. Med. Rev. 1998;3:114–127. [PubMed] [Google Scholar]
- 45.Elkins PA, Ho YS, Smith WW, Janson CA, D'Alessio KJ, McQueney MS, Cummings MD, Romanic AM. Structure of the C-terminally truncated human proMMP9, a gelatin-binding matrix metalloproteinase. Acta Crystallogr. Sect. D. 2002;58:1182–1192. doi: 10.1107/s0907444902007849. [DOI] [PubMed] [Google Scholar]
- 46.Maseras F, Morokuma K. IMOMM: a new integrated ab initio + molecular mechanics geometry optimization scheme of equilibrium structures and transition states. J. Comp. Chem. 1995;16:1170–1179. [Google Scholar]
- 47.DeLano WL. The PyMol molecular graphics system. DeLano Sci.; San Carlos, CA: 2002. http://www.pymol.org. [Google Scholar]