

In addition to spellchecking during DNA replication and modulating recombination, DNA mismatch repair (MMR) promotes cytotoxic responses to certain DNA-damaging agents [1]. In mammalian cells, the best-studied response is to Sn1-type methylating agents, including N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) [1]. Notably, MMR-deficient mammalian cells are resistant to the cytotoxic effects of these agents. A recent report showed that MMR deficiency conferred resistance to MNNG in yeast cells crippled for both homologous recombination (rad52Δ) and the detoxifying enzyme methylguanine methyltransferase (mgt1Δ) [2]. To better understand the response, we searched for additional genes modulating sensitivity to MNNG in rad52Δ mgt1Δ budding yeast. In addition to alleles of known MMR genes, we isolated an allele of DCD1 encoding the enzyme deoxycytidylate deaminase, which influences the dCTP:dTTP nucleotide pool ratio by catalyzing the conversion of dCMP to dUMP [3]. Models of the MMR-dependent cytotoxic response to Sn1-type methylating agents have included the incorporation of dTTP opposite O6-methyl guanine (O6metG) in the template [1]. Our findings lend further support to this aspect of the MMR-dependent response and highlight a mechanism for ‘methylation’ resistance that may be of therapeutic relevance for human cancer.
To better understand the response of budding yeast to DNA methylation damage, we mutagenized a rad52Δ mgt1Δ strain to ~33% survival with ethyl methanesulfonate, and screened for mutants resistant to MNNG. After screening ~10,000 colonies, 18 colonies repeatedly tested resistant. In appropriate crosses, one-half of the rad52Δ mgt1Δ segregants were MNNG resistant, suggesting that a single gene mutation was responsible for the resistance trait and that the mutation was unlinked to either RAD52 or MGT1 (data not shown). Crosses to a rad52Δ mgt1Δ strain produced diploids that were each sensitive to MNNG, indicating that all 18 MNNGr mutations were recessive. Next, we performed complementation tests amongst the mutant collection and with MMR genes that, when mutated, have been found to confer resistance to MNNG, i.e. mlh1Δ, msh2Δ, pms1Δ, msh6Δ ([2] and our unpublished data). Not surprisingly, complementation tests suggested that we had isolated multiple alleles of MLH1 (6), MSH2 (2), PMS1 (3) and MSH6 (6). However, one recessive mutation defined a separate complementation group, initially designated drm1-1 (damage response to methylation).
To identify, by complementation, the gene associated with the MNNG resistance, we transformed the drm1-1 strain with a centromere-based yeast genomic library. Among ~20,000 transformants screened for MNNG sensitivity, two complemented colonies were identified and the library clones isolated. Sequencing revealed that these clones harbored identical genomic inserts containing seven potential open reading frames, including the DCD1 gene. We sequenced the DCD1 gene in the MNNG-resistant (rad52Δ mgt1Δ) strain and detected a mutation (G to T) predicted to cause a serine to phenylalanine change at residue 178, a residue conserved in the human deoxycytidylate deaminase gene Dctd1 (Figure S1 in Supplemental Data).
To further substantiate that the S178F change in Dcd1 was responsible for MNNG resistance in the drm1-1 strain, we introduced the dcd1-S178F mutation into the genome of the wild-type W303 strain. Because DCD1 is non-essential, we also constructed a dcd1Δ strain. The dcd1-S178F and dcd1Δ strains were each crossed with a rad52Δ mgt1Δ strain and haploid, double and triple mutant strains were generated. When compared with the rad52Δ mgt1Δ ‘parental’ strain, the rad52Δ mgt1Δ dcd1-S178F and rad52Δ mgt1Δ dcd1Δ strains showed an increased level of MNNG resistance that was statistically indistinguishable from both the rad52Δ mgt1Δ pms1Δ and rad52Δ mgt1Δ mlh1Δ strains (Figure 1A and Figure S2 in Supplemental Data). In addition, dcd1 deficiency in wild-type (RAD52 MGT1) cells did not confer detectable resistance to MNNG. Taken together with the LD50 values of each strain (see Figure 1 legend), our results show that an inactivating mutation of DCD1 in rad52Δ mgt1Δ cells confers resistance to Sn1-type DNA methylation damage.
Figure 1.
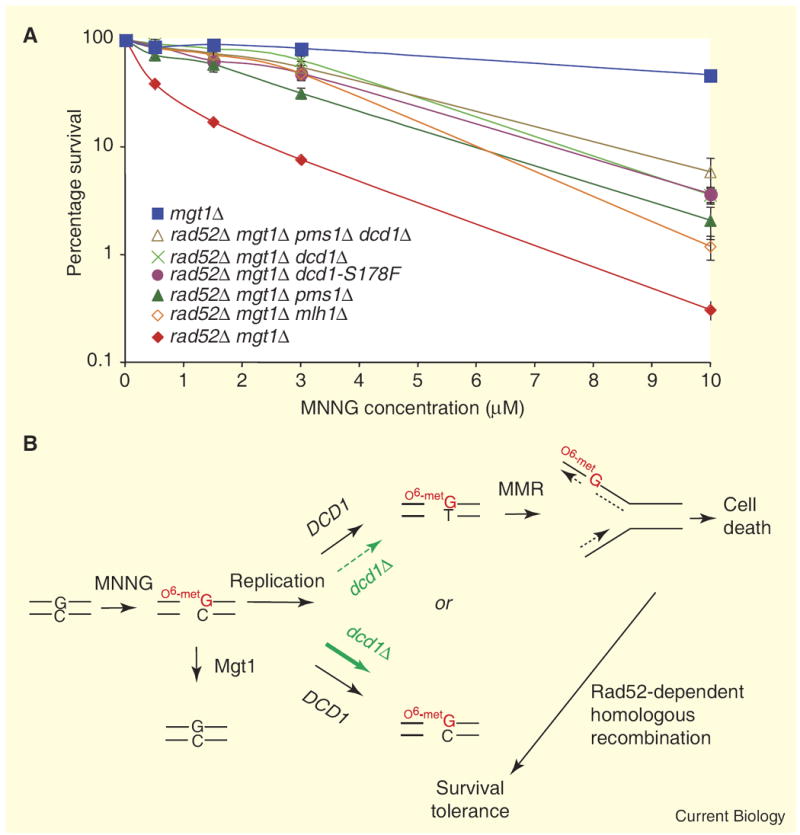
(A) MNNG survival curves: strains were exposed for 45 min to increasing amounts of MNNG. Error bars represent standard error of the mean. The LD50 values for mgt1Δ, rad52Δ mgt1Δ dcd1Δ pms1Δ, rad52Δ mgt1Δ dcd1Δ, rad52Δ mgt1Δ dcd1-S178F, rad52Δ mgt1Δ pms1Δ, rad52Δ mgt1Δ mlh1Δ and rad52Δ mgt1Δ strains were 9.7 μM, 2.4 μM, 2.2 μM, 2.1 μM, 1.8 μM, 1.6 μM and 0.25 μM MNNG, respectively. (B) A model for the cellular response to MNNG treatment in budding yeast. The scheme is modified from a previous report [2] to include the role of DCD1, which, when absent, results in reduced O6metG/T mispair formation following replication, reduced MMR-dependent processing and resistance to cell death. With the exception of the ‘heavy’ and ‘dashed’ arrows signifying consequences in the dcd1Δ strain, the model applies to wild-type yeast. Further, following replication, remaining O6metG/C residues will either be detoxified by the methyltransferase or diluted out by subsequent cell cycles. Although as shown the model infers MMR-dependent ‘futile repair’, ‘direct signaling’ must be considered (for reviews, see [1,12]). Support for the futile cycling model comes from several studies including in vitro results showing reiterative MMR excision cycles on O6metG/T mismatch-containing plasmid substrates [13]. Support for direct signaling comes from a ‘separation-of-function’ allele of mouse Msh6 that compromises spellchecking without affecting the DNA-damage response [14] and from studies showing that MutSα–MutLα complexes are required for ATR–ATRIP signaling from methylation damage sites [15].
As reported previously, DCD1 deficiency in yeast resulted in elevation of dCTP pools and a slight decrease in dTTP pools [4] and reduced the frequency of spontaneous [4] and MNNG-induced [5] G/C to A/T mutations. To address possible yeast strain background effects in our study, we measured dNTP pool levels in the most relevant strains used, as described [6]. Consistent with a previous study [4], dcd1Δ in our strain background also resulted in elevated levels of dCTP and slightly diminished dTTP pools (see Table 1). Notably, MMR deficiency (mlh1Δ) had no significant effect on dNTP pools. The MMR-dependent cytotoxic response to Sn1-type methylating agents generally is considered to be dependent on the formation of O6metG/T mispairs during replication [1]. Because of the increased dCTP: dTTP ratio due to dcd1Δ, the likelihood of incorporation of T opposite O6metG by polymerase is predicted to be greatly diminished. Therefore, we reasoned that MNNG treatment of dcd1Δ cells resulted in fewer O6metG/T mispairs, less involvement by MMR, and cytotoxic resistance/tolerance, assuming that DCD1 and MMR act in the same methylation damage response pathway. To test this notion, we compared the MNNG responses of rad52Δ mgt1Δ dcd1Δ and rad52Δ mgt1Δ pms1Δ dcd1Δ strains: the quadruple mutant appeared no more resistant to MNNG than the triple mutant (Figure 1A,B; see legend for LD50 values). These results support the idea that in yeast the formation of O6metG/T mispairs during replication is required for the observed MMR-dependent cytotoxicity of MNNG. Shown in Figure 1B is a schematic incorporating the DCD1-related findings reported here and those from a previous report [2].
Table 1.
Measurements of deoxynucleotide pools.
| Strain | dNTPs, pmol/106 cells
|
|||
|---|---|---|---|---|
| dATP | dTTP | dCTP | dGTP | |
| rad52Δ mgt1Δ | 1.33 ± 0.23 | 4.39 ± 0.96 | 2.02 ± 0.22 | 0.87 ± 0.22 |
| rad52Δ mgt1Δ mlh1Δ | 1.74 ± 0.24 | 4.92 ± 0.88 | 2.84 ± 0.16 | 1.13 ± 0.14 |
| rad52Δ mgt1Δ dcd1Δ | 1.68 ± 0.36 | 2.14 ± 0.30 | 7.49 ± 0.59 | 0.73 ± 0.17 |
Using mid-log phase yeast cultures grown in rich medium, dNTP pools were determined in duplicate on each of three separate extractions.
The MMR-dependent response to Sn1-type methylating agents most likely involves additional proteins besides those normally associated with MMR-dependent spellchecking (for a review, see [1]). Our study has uncovered one such protein, DCD1, which modulates dCTP:dTTP pool levels and therefore influences sensitivity to agents that induce formation of O6metG. Several studies with cultured rodent cells suggested that Dctd deficiency increased the dCTP:dTTP pool ratio [7-9]. However, we are not aware of any isogenic pairs of proficient/deficient cell lines to test rigorously the role of Dctd in the response to methylation damage. Of interest, however, Meuth [7] showed that elevated dTTP pools increased sensitivity to MNNG in Chinese hamster ovary cells and speculated that misincorporation of thymine opposite O6metG was the basis for the observed toxicity.
Finally, our findings may also have relevance to cancer chemotherapy. For example, reduced DCTD levels in a tumor might compromise the clinical response to the Sn1-type methylation agent temozolomide or the purine analog 6-mercatopurine, used in the treatment of glioblastoma multiforme [10] and certain hematological malignancies [11], respectively.
Supplemental data
Supplemental data are available at http://www.current-biology.com/cgi/content/full/17/17/R755/DC1
Acknowledgments
We thank Andrew Buermeyer, Jennifer Johnson, Ashleigh Miller and Sandra Dudley for critical reading of the manuscript. This work was supported by U.S. Army Research Office Grant No. W911NF-06-1-0110 to C.K.M. and by National Institutes of Health grant 5R01 GM45413 to R.M.L.
References
- 1.Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 2.Cejka P, Mojas N, Gillet L, Schar P, Jiricny J. Homologous recombination rescues mismatch-repair-dependent cytotoxicity of S(N)1-type methylating agents in S. cerevisiae. Curr Biol. 2005;15:1395–1400. doi: 10.1016/j.cub.2005.07.032. [DOI] [PubMed] [Google Scholar]
- 3.McIntosh EM, Haynes RH. Isolation of a Saccharomyces cerevisiae mutant strain deficient in deoxycytidylate deaminase activity and partial characterization of the enzyme. J Bacteriol. 1984;158:644–649. doi: 10.1128/jb.158.2.644-649.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kohalmi SE, Glattke M, McIntosh EM, Kunz BA. Mutational specificity of DNA precursor pool imbalances in yeast arising from deoxycytidylate deaminase deficiency or treatment with thymidylate. J Mol Biol. 1991;220:933–946. doi: 10.1016/0022-2836(91)90364-c. [DOI] [PubMed] [Google Scholar]
- 5.Kunz BA, Henson ES, Karthikeyan R, Kuschak T, McQueen SA, Scott CA, Xiao W. Defects in base excision repair combined with elevated intracellular dCTP levels dramatically reduce mutation induction in yeast by ethyl methanesulfonate and N-methyl-N’-nitro-N-nitrosoguanidine. Environ Mol Mutagen. 1998;32:173–178. [PubMed] [Google Scholar]
- 6.Muller EG. Deoxyribonucleotides are maintained at normal levels in a yeast thioredoxin mutant defective in DNA synthesis. J Biol Chem. 1994;269:24466–24471. [PubMed] [Google Scholar]
- 7.Meuth M. Role of deoxynucleoside triphosphate pools in the cytotoxic and mutagenic effects of DNA alkylating agents. Somatic Cell Genet. 1981;7:89–102. doi: 10.1007/BF01544750. [DOI] [PubMed] [Google Scholar]
- 8.Weinberg G, Ullman B, Martin DW., Jr Mutator phenotypes in mammalian cell mutants with distinct biochemical defects and abnormal deoxyribonucleoside triphosphate pools. Proc Natl Acad Sci USA. 1981;78:2447–2451. doi: 10.1073/pnas.78.4.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dare E, Zhang LH, Jenssen D, Bianchi V. Molecular analysis of mutations in the hprt gene of V79 hamster fibroblasts: effects of imbalances in the dCTP, dGTP and dTTP pools. J Mol Biol. 1995;252:514–521. doi: 10.1006/jmbi.1995.0516. [DOI] [PubMed] [Google Scholar]
- 10.Robins HI, Chang S, Butowski N, Mehta M. Therapeutic advances for glioblastoma multiforme: current status and future prospects. Curr Oncol Rep. 2007;9:66–70. doi: 10.1007/BF02951428. [DOI] [PubMed] [Google Scholar]
- 11.Karran P, Offman J, Bignami M. Human mismatch repair, drug-induced DNA damage, and secondary cancer. Biochimie. 2003;85:1149–1160. doi: 10.1016/j.biochi.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 12.Wang JY, Edelmann W. Mismatch repair proteins as sensors of alkylation DNA damage. Cancer Cell. 2006;9:417–418. doi: 10.1016/j.ccr.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 13.York SJ, Modrich P. Mismatch repair-dependent iterative excision at irreparable O6-methylguanine lesions in human nuclear extracts. J Biol Chem. 2006;281:22674–22683. doi: 10.1074/jbc.M603667200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang G, Scherer SJ, Shell SS, Yang K, Kim M, Lipkin M, et al. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell. 2004;6:139–150. doi: 10.1016/j.ccr.2004.06.024. [DOI] [PubMed] [Google Scholar]
- 15.Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell. 2006;22:501–510. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental data are available at http://www.current-biology.com/cgi/content/full/17/17/R755/DC1