

Abstract
A novel fungal metabolite, apicidin [cyclo(N-O-methyl-l-tryptophanyl-l-isoleucinyl-d-pipecolinyl-l-2-amino-8-oxodecanoyl)], that exhibits potent, broad spectrum antiprotozoal activity in vitro against Apicomplexan parasites has been identified. It is also orally and parenterally active in vivo against Plasmodium berghei malaria in mice. Many Apicomplexan parasites cause serious, life-threatening human and animal diseases, such as malaria, cryptosporidiosis, toxoplasmosis, and coccidiosis, and new therapeutic agents are urgently needed. Apicidin’s antiparasitic activity appears to be due to low nanomolar inhibition of Apicomplexan histone deacetylase (HDA), which induces hyperacetylation of histones in treated parasites. The acetylation–deacetylation of histones is a thought to play a central role in transcriptional control in eukaryotic cells. Other known HDA inhibitors were also evaluated and found to possess antiparasitic activity, suggesting that HDA is an attractive target for the development of novel antiparasitic agents.
Keywords: cyclic tetrapeptide, Apicomplexa, antiparasitic, malaria, coccidiosis
Protozoan parasites of the subphylum Apicomplexa remain significant threats to human and animal health worldwide. With respect to human health, malaria remains one of the leading causes of death in the world, resulting in the loss of over 1.5 million lives per year (1). Widespread multidrug resistance to malaria has developed, and few, if any, new therapeutic agents will be available in the foreseeable future. Another Apicomplexan parasite, Cryptosporidium parvum, was recently identified by the World Health Organization as an emerging global health problem (2). The rapid spread of cryptosporidiosis has been reported in urban slums (3), and there have been several major water-borne outbreaks in developed countries in which thousands of individuals were infected (4). In immune compromised individuals, such as AIDS patients, Cr. parvum infections are incurable and lead to chronic diarrhea and wasting disease. Despite its medical importance, there is currently no therapy for treating cryptosporidiosis. Another important apicomplexan infection in immune-compromised patients is Toxoplasma gondii, which is becoming a relatively common problem in AIDS patients (5). Although methods of treating toxoplasmosis exist, better therapeutic agents are clearly needed.
In animal health, the Apicomplexan parasites cause major economic losses in livestock and poultry throughout the world. Eimeria parasites are responsible for coccidiosis in poultry and many other domesticated animals. Infection of the gut epithelium by these intracellular parasites results in severe morbidity and mortality, particularly in chickens. Poultry producers worldwide routinely employ chemical prophylaxis to prevent serious coccidiosis outbreaks. Resistance to currently available coccidiostats is prevalent, and new anticoccidial agents are needed. T. gondii is an important cause of abortion and morbidity in livestock, especially sheep and goats (6), and species of Cryptosporidium cause widespread and rapidly transmitted diarrheal illness in several mammalian hosts, especially calves, neonatal lambs and goats, and young foals (7).
In this paper, a novel natural product, apicidin [cyclo(N-O-methyl-l-tryptophanyl-l-isoleucinyl-d-pipecolinyl-l-2-amino-8-oxodecanoyl)], that has broad spectrum activity against the Apicomplexan parasites is described, and experimental evidence that demonstrates that this compound kills parasites by inhibiting histone deacetylase (HDA), a key nuclear enzyme involved in transcriptional control, is provided.
MATERIALS AND METHODS
Source of Compounds and Organisms.
[3H]Apicidin A (2-N-desmethoxy[3H]apicidin, specific activity 18.7 mCi/mg; 1 Ci = 37 GBq), Ac-Gly-Ala-Lys(ɛ-[3H]Ac)-Arg-His-Arg-Lys(ɛ-[3H]Ac)-Val-NH2 (specific activity 3.8 Ci/mmol), β-hydroxy-HC-toxin, and trichostatin were prepared at Merck Research Laboratories, Rahway, NJ. Sodium [14C]acetate (60 mCi/mmol) was purchased from Amersham. Sodium butyrate and HC-toxin were from Sigma. Organisms for in vitro studies were obtained from a variety of sources: Plasmodium berghei (strain KBG 173), A. Ager (University of Miami, Miami); Plasmodium falciparum (Dd2 strain), D. Chakraborti (University of Florida, Gainesville, FL); Neospora caninum (strain NC-1-2C) and Caryospora bigenetica, D. Lindsay and C. Sundermann (Auburn University, Auburn, AL). Human blood products were from the North Jersey Blood Center.
Determination of in Vitro Antiprotozoal Activity.
Conditions for the in vitro culture of parasites and determination of minimal inhibitory concentrations [defined as the lowest concentration (nanograms per milliliter) at which parasite growth was fully inhibited] for compounds were conducted according to previously described methods. For Eimeria tenella, the 48-hr assay as described by Schmatz et al. (8) was used; for T. gondii, Besnoitia jellisoni, and N. caninum, the method of Roos et al. (9) was used; for Ca. bigenetica, the 7-day assay as described by Sundermann et al. (10) was used; for P. falciparum [chloroquine-resistant strain Dd2, grown according to Trager and Jensen (11)], drug sensitivity was determined over 48 hr visually by light microscopy of stained blood smears; and activity against Cr. parvum was determined according to Woods et al. (12) with rat serum at a 1:1000 dilution. Test compounds were dissolved in 100% dimethyl sulfoxide (DMSO), and working dilutions were prepared in growth media to deliver a final concentration of compound in ≤0.5% (vol/vol) final DMSO concentration.
Mouse Malaria Studies.
An acute strain of P. berghei (KBG 173) was maintained by routine passage in BALB/C mice. To evaluate compounds, BALB/c mice (female, 20–22 g) were injected i.p. with 106 infected erythrocytes obtained from mice with acute infections. This dose routinely resulted in 100% mortality of control mice at 6–10 days post infection (p.i.). All treatments were initiated 2 hr p.i. Compounds were dissolved in 100% DMSO and diluted to the required concentrations in 10% DMSO/90% mouse serum. Groups of five infected mice each were dosed twice a day orally by gavage (0.25 ml) or by i.p. injection (0.2 ml) for 3 consecutive days. Control mice were treated for the same period with vehicle alone. The percent infected erythrocytes per 1000 cells was determined on day 6 p.i. by microscopic examination of thin smears of venous blood.
E. tenella Parasites.
Chickens were infected orally with 7.5 × 104 E. tenella LS18 sporulated oocysts. The unsporulated oocysts were harvested from the ceca 7 days p.i. and purified according to the method of Schmatz et al. (13), and then sporulated by continual agitation for 36 hr at 29°C.
Apicidin A Binding Assay.
Soluble extracts for binding studies were prepared by vortexing 2 × 109 E. tenella sporulated oocysts with 4-mm glass beads (4 ml) and 50 mM Hepes (pH 7.4) containing 0.1 mM phenylmethylsulfonyl fluoride (5 ml) for 20 min. The resulting homogenate was centrifuged (100,000 × g, 1 h), and the supernatant (S100) was used directly. Test compounds were added to assay tubes containing 1.3 ng/ml [3H]apicidin A, 950 μl of buffer A (50 mM Hepes, pH 7.4/0.02% Triton X-100), and 200 μg of E. tenella S100 (50 μl). Samples were incubated for 1 hr at 25°C and filtered through Whatman GF/B glass fiber filters (presoaked in 0.6% polyethyleneimine for 1 hr at 25°C), and the filters were washed with buffer A (3 × 2.0 ml at 4°C) by vacuum filtration and dried. The radioactivity associated with filters was determined by scintillation counting using Ready-SAFE (Amersham).
HDA Enzyme Assay.
Soluble extracts of HDA were prepared by vortexing 1 × 109 E. tenella unsporulated oocysts with glass beads (5 ml; 0.3–3.0 mm in diameter) and 2 ml of buffer B (25 mM Hepes-Na, pH 7.4, with 1.0 mM MgCl2) for 7 min. The homogenate was removed, the beads were washed with buffer B (10 ml), and the pooled homogenate was centrifuged (at 100,000 × g for 0.5 hr). The pellet was resuspended in buffer B (4 ml), incubated at 4°C for 19 hr, and centrifuged (at 100,000 × g for 0.5 hr). The supernatant was concentrated (centricon 10) to yield an HDA extract (1.5 ml/8.3 mg/ml protein). The standard assay (200 μl) contained 10 mM Hepes-Na (pH 7.4), 1 M sucrose, 0.1 mg/ml BSA, 0.01% (vol/vol) Triton X-100, 0.5% (vol/vol) DMSO, 280 nM Ac-Gly-Ala-Lys(ɛ-[3H]Ac)-Arg-His-Arg-Lys(ɛ-[3H]Ac)-Val-NH2 (specific activity 3.8 Ci/mmol; ≈0.05 × Km), and E. tenella extract (0.42 mg/ml) with or without inhibitor (delivered in 100% DMSO). The reaction mixture, minus substrate, was preincubated at 25°C for 30 min. The substrate was then added, and the mixture was incubated for a further 30 min at 25°C and then stopped by addition of a solution of 0.1 M acetic acid/0.5 M hydrochloric acid (20 μl). Released [3H]acetate was extracted with ethyl acetate (1 ml), an aliquot of the ethyl acetate layer (0.9 ml) was removed, and mixed with Aquasol 2 scintillation cocktail (6 ml), and the radioactivity was counted. Uninhibited HDA-catalyzed product formation (≈1500 dpm) was calculated by subtracting the radioactivity obtained in zero enzyme controls.
Histone Hyperacetylation Assay.
Approximately 5 × 108 P. falciparum-infected erythrocytes were preincubated in RPMI 1640 medium with 10% A+ human serum with test compounds for 2 hr at 37°C. The infected erythrocytes were then centrifuged (at 1000 × g for 5 min), resuspended in RPMI 1640 medium containing 250 μCi sodium [14C]acetate (60 mCi/mmol; Amersham) and the same level of test compounds as in the pretreatment, and incubated an additional 2 hr at 37°C. The cells were then washed three times in PBS, a parasite nuclear fraction was prepared (14), and protein was precipitated with 0.4 M H2SO4. Acid soluble proteins were then precipitated with acetone and fractionated by electrophoresis on acid urea triton (AUT) gels according to Alfageme et al. (15) with modifications as described by Lennox and Cohen (16). Gels were fixed in methanol/acetic acid for 16 hr, treated with Enlighten (NEN) for 30 min, dried, and exposed to film (Fuji RX medical x-ray film) at −70°C.
RESULTS AND DISCUSSION
Apicidin (see Fig. 1, 1) is a novel cyclic tetrapeptide isolated from fermentations of Fusarium spp. (ATCC 74289; ATCC 74322) obtained from Costa Rica. It irreversibly prevents the in vitro development of intracellular Apicomplexan parasites at low nanograms per milliliter levels (Table 1). Apicidin represents a new antiprotozoal agent that operates through a unique mode of action against drug-resistant human malaria in vitro (Table 1). Furthermore, apicidin demonstrates parenteral and oral efficacy against an acute strain of P. berghei in mice (see Fig. 2). The potential of apicidin as a broad spectrum antiprotozoal is exemplified by its potent in vitro activity against Cr. parvum, an invasive opportunistic pathogen in humans and animals, for which there is currently no therapy (17). The antiparasitic spectrum of apicidin is unique in that it appears specific for the Apicomplexans and is inactive against several of the flagellated protozoa, including Giardia lamblia, Tritrichomonas foetus, Leishmania major, Trypanosoma cruzi, and Trypanosoma brucei (data not shown).
Figure 1.
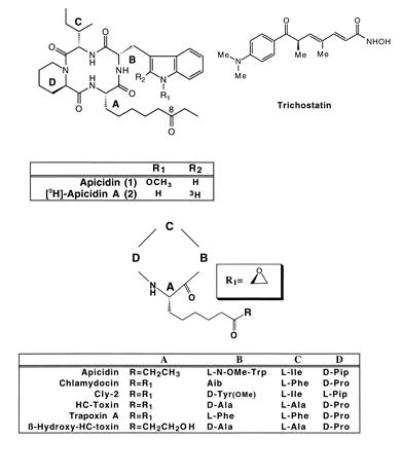
(A and B) The structures of apicidin and trichostatin (A), and structurally related tetrapeptides (B). Pip, Pipecolic acid; Aib, aminoisobutyric acid.
Table 1.
In vitro antiprotozoal efficacy of apicidin and related compounds
| Parasite | Minimal inhibitory concentration, ng/ml
|
|||
|---|---|---|---|---|
| Apicidin | Apicidin A | HC-toxin | β-Hydroxy HC-toxin | |
| E. tenella | 62 | 31 | 16 | >1000 |
| T. gondii | 8 | 15 | 15 | >1000 |
| P. falciparum | 125 | 125 | 31 | >1000 |
| Cr. parvum | 30 | 70 | 180 | >1000 |
| N. caninum | 15 | 30 | 15 | >1000 |
| B. jellisoni | 4 | 1 | 8 | >1000 |
| Ca. bigenetica | 8 | — | — | — |
In vitro minimal inhibitory concentrations for apicidin and other compounds against a panel of Apicomplexan parasites were determined as described in Materials and Methods.
Figure 2.
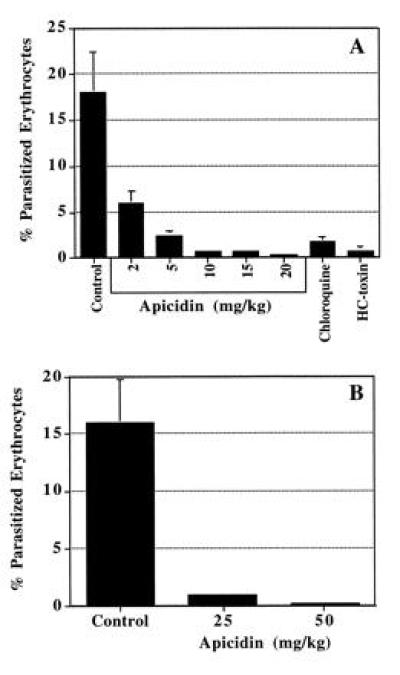
Evaluation of apicidin and HC-toxin against acute P. berghei malaria. BALB/C mice were injected i.p. with 106 P. berghei-infected erythrocytes and then treated twice a day for 3 days with HC-toxin or apicidin at various doses starting at 2 hr p.i. Results are expressed as the percent of parasitized erythrocytes at 6 days p.i. (A) Parenteral (i.p.) treatment with HC-toxin (50 mg/kg), apicidin, or chloroquine (10 mg/kg). (B) Oral treatment with apicidin. Error bars represent SEM. Each group consisted of five mice.
Several naturally occurring cyclic tetrapeptides have been reported in the literature and are structurally related to apicidin, including HC-toxin (18), trapoxin A (19), WF-3161 (20), cly-2 (21), and chlamydocin (22) (Fig. 1). These related cyclic tetrapeptides display potent low nanomolar antineoplastic activity, but their antiprotozoal properties are unknown. We have also evaluated HC-toxin for antiprotozoal activity and discovered that it has comparable in vitro and in vivo efficacy to apicidin (Table 1 and Fig. 2A). Additionally, apicidin was found to inhibit mammalian (HeLa) cell proliferation in vitro (50% inhibitory concentrations, 50–100 nM).
The antiproliferative activity of trapoxin A (Fig. 1), has been reported to occur as a result of potent, irreversible inhibition of mammalian HDA (23). In addition, trapoxin A exhibits a variety of biological properties similar to those of another known HDA inhibitor, trichostatin (24), including 10-fold cross-resistance to a trichostatin-resistant cell line at both the cellular and the enzymatic level. More recently, HC-toxin and chlamydocin also have been shown to inhibit partially purified HDA from maize, Physarum, and chicken (25). Because histones play a key role in transcriptional regulation, and the continuous acetylation/deacetylation of the ɛ-amino group of specific histone lysine residues is required for this process, it is believed that the inhibition of histone deacetylation interferes with transcriptional control and thus cell proliferation. Similarities in the biological effects of apicidin to those described for structurally related HDA inhibitors prompted us to explore HDA inhibition as the potential mechanism of action of apicidin against protozoan parasites.
Initially, a binding assay was developed (see Materials and Methods) to identify the molecular target of apicidin and to follow structure–activity relationships of related analogs. [3H]Apicidin A was chosen as the radioligand because it is an equipotent congener/analog of apicidin (Figs. 1 and 2, and Table 1). Sporulated oocysts of the Apicomplexan parasite E. tenella were chosen as the source of parasite extracts because they are available in large quantities and are free from host contamination. Approximately 90% of the specific binding of [3H]apicidin A was present in the 100,000 × g supernatant (S100) from oocyst homogenates, with a Kd of 4 nM. Apicidin, apicidin A, and known HDA inhibitors HC-toxin (25) and trichostatin (24) (Fig. 1) competed with [3H]apicidin A binding with IC50 values in the 4–50 nM range, whereas an inactive analog, β-hydroxy-HC-toxin (Fig. 1), which lacked antiparasitic activity, also lacked binding activity (Tables 1 and 2).
Table 2.
The effects of apicidin and other compounds on [3H]apicidin A binding and HDA activity
| Compound | IC50
|
|
|---|---|---|
| Binding | Enzyme Activity | |
| Apicidin | 4 nM | 0.7 nM |
| Apicidin A | 4 nM | 1.0 nM |
| HC-toxin | 50 nM | 30 nM |
| β-Hydroxy-HC-toxin | >1000 nM | >1000 nM |
| Trichostatin | 40 nM | 3.8 nM |
| Butyrate | 1.5 mM | 38 μM |
[3H]Apicidin A binding and HDA activity in E. tenella homogenates were assayed as described in Materials and Methods.
Apicidin and several known HDA inhibitors were also evaluated as inhibitors of partially purified E. tenella HDA activity. E. tenella HDA catalytic activity was measured as [3H]acetate release from the radiolabeled octapeptide [Ac-Gly-Ala-Lys(ɛ-[3H]Ac)-Arg-His-Arg-Lys(ɛ-[3H]Ac)-Val-NH2], a known substrate of mammalian HDA (26). Apicidin, HC-toxin, trichostatin, and another known HDA inhibitor, sodium butyrate (27), all inhibited the E. tenella HDA activity at levels comparable to those seen in the binding assay (Table 2). β-Hydroxy-HC-toxin, which lacked binding and antiparasitic activities, did not inhibit E. tenella HDA enzyme activity. Enzyme activity and [3H]apicidin A binding activity co-eluted on both gel filtration and ion-exchange chromatographies (data not shown).
Inhibition of mammalian HDA by sodium butyrate (27), trapoxin A (23), and HC-toxin (25) results in the accumulation of hyperacetylated histones. This effect can be visualized by AUT gel electrophoresis of histones extracted from treated cells. Increased acetylation results in decreased mobility of histones in AUT gels, and the appearance of multiple bands corresponding to histones with 1, 2, 3, and 4 acetylated lysine residues. P. falciparum-infected human erythrocytes were used to explore parasite histone hyperacetylation because this system is free of host cell histones. As shown in Fig. 3A, apicidin induces hyperacetylation of parasite histones in a dose-dependent manner. Moreover, apicidin, HC-toxin, and trichostatin all induce hyperacetylation of parasite histones (Fig. 3B) at 100 ng/ml, whereas neither β-Hydroxy-HC-toxin nor the antimalarial agent chloroquine induce hyperacetylation of parasite histones at 500 ng/ml.
Figure 3.
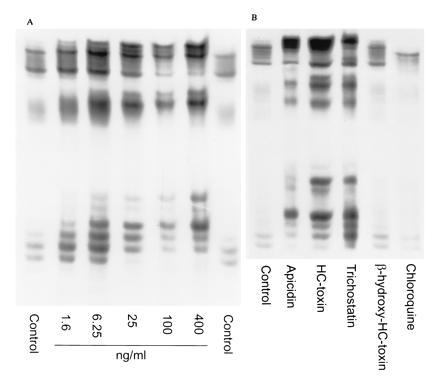
The effect of apicidin and other compounds on histone acetylation in P. falciparum. Treatment of P. falciparum-parasitized erythrocytes with vehicle only, control: 1.6–400 ng/ml apicidin (A), or vehicle only, control; 100 ng/ml of apicidin, HC-toxin, or trichostatin, or 500 ng/ml β-hydroxy-HC-toxin or chloroquine (B). Histones were extracted from parasites and visualized by AUT gel electrophoresis and autoradiography. Labeled species on the AUT gels are identified as histones as (i) they comigrate with HeLa cell histones on SDS/polyacrylamide gels, (ii) they bind antisera raised against acetylated histones (28) (a generous gift from B. Turner, University of Birmingham Medical School, Birmingham, AL), (iii) [14C]acetate incorporation increases as a result of treatment with diverse inhibitors of HDA, and (iv) acetylated histone species show slower rates of migration than nonacetylated histones (28). Uninfected erythrocytes processed under similar conditions did not show any labeled bands on AUT gels (data not shown).
The results presented here clearly show that apicidin as well as the known mammalian HDA inhibitors, HC-toxin and trichostatin, are potent inhibitors of parasite HDA. The 2-amino-8-oxo-decanoic acid moiety of apicidin is unique relative to related tetrapeptides where an epoxide is required for biological activity. The 2-amino-8-oxo-decanoic acid presumably mimics the ɛ-amino acetylated lysine residues of histone substrates, resulting in potent reversible inhibition of HDA. The recent molecular characterization of human HDA (29) and histone acetyltransferase from Tetrahymena (30) has provided novel insights into their roles in the regulation of chromatin structure and gene transcription in eukaryotes. Apicidin and its analogs are tools that can be used to dissect the components of the regulatory pathways, mediating these critical processes, and in turn, they will enhance understanding of the intricacies of transcriptional regulation. The antiprotozoal properties of apicidin resulting from inhibition of HDA implies pathways of similar importance and probable complexity exist in protozoa. The enzymatic machinery involved in gene expression in Apicomplexans therefore represents a novel target for antiprotozoal therapy. The antiproliferative activity of apicidin against mammalian cells implies that issues of selectivity of inhibitors for parasite versus host enzyme will need to be addressed, but based on the spectrum of antiparasitic activity displayed by HDA inhibitors and their in vivo efficacy against malaria, HDA appears to be a viable target for the development of antiparasitic agents.
Acknowledgments
We would like to acknowledge the following for their support: the Instituto Nacional de Biodiversidad (InBio), Santa Domingo, Costa Rica, for acquisition of Fusarium spp.; Drs. Dennis Dean, Yui Sing Tang, Avery Rosegay (Merck Research Laboratories, Rahway, NJ) for the [3H]acetate labeling of the HDA octapeptide substrate and the preparation of [3H]apicidin A; Drs. K. Woods and S. Upton (Kansas State University, Manhattan, KS) for performing in vitro studies with Cr. parvum and M. A. Powles (Merck Research Laboratories) for performing in vitro studies with Ca. bigenetica; Dr. A. Ager (University of Miami), Dr. D. Chakraborti (University of Florida, Gainesville, FL), and Drs. D. Lindsay and C. Sundermann (Auburn University, Auburn, AL) for supplying organisms; and the North Jersey Blood Center for providing blood products for maintaining P. falciparum in vitro.
Footnotes
Abbreviations: HDA, histone deacetylase; p.i., post infection; AUT, acid urea triton.
References
- 1.World Health Organization, Malaria Unit. Bull W H O. 1993;71:281–284. [PMC free article] [PubMed] [Google Scholar]
- 2.WHO (1994) New and Re-emerging Infectious Diseases, W. H. O. Fact Sheet No. N97.
- 3.Newman R D, Zu S X, Wuhib T, Lima A A M, Guerrant R L, Sears C L. Ann Intern Med. 1994;120:500–505. doi: 10.7326/0003-4819-120-6-199403150-00009. [DOI] [PubMed] [Google Scholar]
- 4.MacKenzie W R, Hoxie N J, Proctor M E, Gradus M S, Blair K A, Peterson D E, Kazmierczak J J, Addiss D G, Fox K R, Rose J B, Davis J P. N Engl J Med. 1994;331:161–167. doi: 10.1056/NEJM199407213310304. [DOI] [PubMed] [Google Scholar]
- 5.Neva F A, Brown H W, editors. Basic Clinical Parasitology. East Norwalk, CT: Appleton & Lange; 1994. pp. 44–50. [Google Scholar]
- 6.Dubey J P, Beattie C P. Toxoplasmosis of Animals and Man. Boca Raton, FL: CRC; 1988. [Google Scholar]
- 7.Current W L, Blagburn B L. Coccidiosis of Man and Domestic Animals. Boca Raton, FL: CRC; 1990. pp. 155–185. [Google Scholar]
- 8.Schmatz D M, Crane M, Murray P K. J Protozool. 1986;33:109–114. doi: 10.1111/j.1550-7408.1986.tb05568.x. [DOI] [PubMed] [Google Scholar]
- 9.Roos D S, Donald R G K, Morrissette N S, Moulton A L C. Methods Cell Biol. 1994;45:27–63. doi: 10.1016/s0091-679x(08)61845-2. [DOI] [PubMed] [Google Scholar]
- 10.Sundermann C A, Lindsay D S, Tibbs R E, Jr, Bailey M A. J Protozool. 1988;35:465–469. doi: 10.1111/j.1550-7408.1988.tb04131.x. [DOI] [PubMed] [Google Scholar]
- 11.Trager W, Jensen J B. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 12.Woods K M, Nesterenko M V, Upton S J. FEMS Microbiol Lett. 1995;128:89–94. doi: 10.1111/j.1574-6968.1995.tb07505.x. [DOI] [PubMed] [Google Scholar]
- 13.Schmatz D M, Crane M S J, Murray P K. J Protozool. 1984;31:181–183. doi: 10.1111/j.1550-7408.1984.tb04314.x. [DOI] [PubMed] [Google Scholar]
- 14.Cary C, Lamont D, Dalton J P, Doerig C. Parasitol Res. 1994;80:255–258. doi: 10.1007/BF00932684. [DOI] [PubMed] [Google Scholar]
- 15.Alfageme C R, Zweidler A, Mahowald L H, Cohen L H. J Biol Chem. 1974;249:3729–3736. [PubMed] [Google Scholar]
- 16.Lennox R W, Cohen L H. Methods Enzymol. 1989;170:532–549. doi: 10.1016/0076-6879(89)70063-x. [DOI] [PubMed] [Google Scholar]
- 17.St. Georgiev V. Drug Dev Res. 1993;28:445–459. [Google Scholar]
- 18.Liesch J M, Sweeley C C, Staffeld G D, Anderson M S, Weber D J, Scheffer R P. Tetrahedron. 1982;38:45–48. [Google Scholar]
- 19.Itazaki H, Nagashima K, Sugita K, Yoshida H, Kawamura Y, Yasuda Y, Matsumoto K, Ishii K, Uotani N, Nakai H, Terui A, Yoshimatsu S, Ikenishi Y, Nakagawa Y. J Antibiot. 1990;43:1524–1532. doi: 10.7164/antibiotics.43.1524. [DOI] [PubMed] [Google Scholar]
- 20.Umehara K, Nakahara K, Kiyoto S, Iwami M, Okamoto M, Tanaka H, Kohsaka M, Aoki H, Imanaka H. J Antibiot. 1983;36:478–483. doi: 10.7164/antibiotics.36.478. [DOI] [PubMed] [Google Scholar]
- 21.Hirota A, Suzuki A, Aizawa K, Tamura S. Agric Biol Chem. 1973;37:955–956. [Google Scholar]
- 22.Closse A, Hugenin R. Helv Chim Acta. 1974;57:533–545. doi: 10.1002/hlca.19740570306. [DOI] [PubMed] [Google Scholar]
- 23.Kijima M, Yoshida M, Sugita K, Horinouchi S, Beppu T. J Biol Chem. 1993;268:22429–22435. [PubMed] [Google Scholar]
- 24.Yoshida M, Kijima M, Akita M, Beppu T. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 25.Brosch G, Ransom R, Lechner T, Walton J D, Loidl P. Plant Cell. 1995;7:1941–1950. doi: 10.1105/tpc.7.11.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kervabon A, Mery J, Parello J. FEBS Lett. 1979;106:93–96. doi: 10.1016/0014-5793(79)80702-4. [DOI] [PubMed] [Google Scholar]
- 27.Riggs M G, Whittaker R G, Neuman J R, Ingram V M. Nature (London) 1977;268:462–464. doi: 10.1038/268462a0. [DOI] [PubMed] [Google Scholar]
- 28.Turner B M, O’Neil L P, Allan I M. FEBS Microbiol Lett. 1989;253:141–145. doi: 10.1016/0014-5793(89)80947-0. [DOI] [PubMed] [Google Scholar]
- 29.Taunton J, Hassig C A, Schreiber S L. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 30.Brownell J E, Zhou J, Ranalli T, Kobayashi R, Edmondson D G, Roth S Y, Allis C D. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]