

Abstract
The expression of a number of human paired box-containing (PAX) genes has been correlated with various types of tumors. Novel fusion genes encoding chimeric fusion proteins have been found in the pediatric malignant tumor alveolar rhabdomyosarcoma (RMS). They are generated by two chromosomal translocations t(2;13) and t(1;13) juxtaposing PAX3 or PAX7, respectively, with a forkhead domain gene FKHR. Here we describe that specific down-regulation of the t(2;13) translocation product in alveolar RMS cells by antisense oligonucleotides results in reduced cellular viability. Cells of embryonal RMS, the other major histiotype of this tumor, were found to express either wild type PAX3 or PAX7 at elevated levels when compared with primary human myoblasts. Treatment of corresponding embryonal RMS cells with antisense olignucleotides directed against the mRNA translational start site of either one of these two transcription factors similarly triggers cell death, which is most likely due to induction of apoptosis. Retroviral mediated ectopic expression of mouse Pax3 in a PAX7 expressing embryonal RMS cell line could partially rescue antisense induced apoptosis. These data suggest that the PAX3/FKHR fusion gene and wild-type PAX genes play a causative role in the formation of RMS and presumably other tumor types, possibly by suppressing the apoptotic program that would normally eliminate these cells.
Pax proteins represent a family of transcriptional regulators expressed mainly during early development. Members of this family play a fundamental role in the regulation of developmental processes (1). Specific human and mouse mutations have revealed critical regulatory functions for several Pax gene products (for review, see ref. 2). Interestingly, the phenotypes of these loss-of-function mutations almost exclusively unveil lack of specific structures, e.g., for Pax2 absence of kidneys, ureters, and genital tracts (3), for Pax5 lack of mature B cells (4), and for Pax6 lack of eye structures (5). Characteristically, these mutations are semidominant in nature, suggesting that the dosage is critical for Pax protein function. Pax3 and Pax7 are normally expressed during mouse embryonal development mainly in specific parts of the neural tube, sensory organs, and in muscle precursor cells in the dermomyotome of the somites. Both factors are down-regulated upon muscle cell differentiation, where expression of Pax7 persists slightly longer that that of Pax3. Interestingly, Pax3 appears to be required for the migration of muscle precursor cells and hence for proper development of limb musculature, suggesting a critical role for this transcription factor during myogenesis (6, 7).
Rhabdomyosarcomas (RMSs) are pediatric soft tissue tumors of skeletal muscle origin. Histologically, two major subtypes can be distinguished: (i) the more prevalent embryonal RMS (eRMS) and (ii) the more aggressive alveolar RMS (aRMS). On the molecular level, aRMS is characterized by a specific chromosomal t(2;13)(q35;q14) translocation that involves the PAX3 and forkhead domain (FKHR) genes (8, 9). This translocation gives rise to a novel chimeric fusion protein between the 5′ region of PAX3 with its intact DNA binding domains and the 3′ region of the FKHR gene product comprising its transactivation domain. In some cases the variant t(1;13)(p36;q14) is observed, where the PAX7 gene is incidentally fused to the same FKHR gene (10). The PAX3/FKHR fusion protein is a more potent transcriptional activator than wild-type PAX3 (11, 12) suggesting enhanced activation of wild-type PAX3 target genes. The specific association of the t(2;13) translocation product with aRMS strongly suggests that this chimeric protein plays a causative role in the development of the tumor.
Using an antisense oligonucleotide strategy, we present evidence that specific down-regulation of the PAX3/FKHR fusion protein can trigger physiological tumor cell death. Similarly, induction of cell death was observed after down-regulation of the wild-type PAX3 or PAX7 proteins, suggesting that PAX gene products might, in these embryonal cancer cells, be required for cell survival.
MATERIALS AND METHODS
Cell Lines.
aRMS cells Rh30 and eRMS cells Rh1 were a generous gift from P. Houghton (St. Jude Children’s Research Hospital, Memphis); RD cells were obtained from American Type Culture Collection. All cells were maintained in DME supplemented with 10% fetal calf serum (GIBCO/BRL).
Oligonucleotide (ODN) Incubations.
For ODN treatments, 105 cells (2 × 105 cells for Rh1) were plated on a 3-mm dish and washed twice with DME without serum 24 hr later. Appropriate ODN concentrations were then resuspended in serum free medium (1 ml per dish), lipofectin (Life Technologies, Grand Island, NY; 10 μg/ml) was added, and the cells were incubated with the mixture for 24 hr. Fresh medium with 10% fetal calf serum was added and changed daily. For counting with a haemocytometer, cells were collected by trypsinization. Cell viability was assessed by trypan blue exclusion. ODN were synthesized as standard phosphodiester molecules on a Gene Assembler (Pharmacia), purified with a Sephadex G-25 column, and resuspended in PBS. ODN integrity after synthesis was assessed by PAGE. Sequences for PAX3 ODN corresponding to nucleotides 123–141 of the human PAX3 cDNA (region of 100% identity with mouse Pax3) are as follows: antisense, 5′-GCGTGGTCATCCTGGGGGC-3′; sense, 5′-GCCCCCAGGATGACCACGC-3′; mutated, 5′-GCGAGGACAACGAGGGGGC-3′ (mutated bases underlined); and scrambled, 5′-AGGGGTCGTGCGCGGTCTG-3′; Sequences for PAX7 ODN corresponding to nucleotides 111–127 of the human PAX7 cDNA are as follows: antisense, 5′-AGGGCCGCCATTCTTGC-3′; sense, 5′-GCAAGAATGGCGGCCCT-3′; and mutated (ms-P3), 5′-AGGGGCCCCTTTCTAGC-3′ (mutated bases underlined).
Detection of PAX3/FKHR Fusion Protein.
Western blot analysis was performed according to standard methods. An equal number of attached, viable cells were lysed directly in SDS sample buffer. Cell lysates were analyzed on a SDS/10% polyacrylamide/taurine gel and transferred to poly(vinylidene difluoride) membranes (Pall). Blots were probed with a polyclonal Pax3 antibody (diluted 1:5000) whose specificity for the PAX3/FKHR fusion protein has been described (11). Protein bands were visualized by means of the Western-Light Chemiluminescent Detection system (Tropix, Bedford, MA), using an alkaline phosphatase-conjugated secondary antibody (Promega). Exposure time was 10 min. Silver staining of a duplicate gel was performed with the Silver Stain kit (Pharmacia).
Expression of PAX mRNA.
RNase protection analysis was performed as described previously except that total RNA was used instead of poly(A)+ RNA (RPAII kit, Ambion, Austin, TX) (13).
Analysis of Apoptosis.
Fluorescence-activated cell sorter analysis was performed after propidium iodine labeling (Cycletest, Becton Dickinson) with a FACScan instrument (Becton Dickinson). All cells in a dish, including the floating cells, were collected for staining. Analysis of 10,000 events was performed using the sfit software (Becton Dickinson). The terminal transferase-mediated dUTP nick-end labeling (TUNEL) assay was performed with the In Situ Cell Death Detection kit, fluorescein (Boehringer Mannheim) according to the manufacturer’s instructions. Micrographs were taken with a Zeiss Axioskop.
Retroviral Infection.
The complete coding region of the murine Pax3 cDNA (13) was cloned into the BamHI site of the retroviral vector pDOL (14) via blunt-end ligation. pDOL with a cDNA insert coding for β-galactosidase (pBAG) was used as a control (15). The vectors were transfected by calcium-phosphate precipitation into the 293 derived amphotrophic packaging cell line BING (ATCC 11554) and supernatant containing retroviral particles collected after 48 hr. Rh1 cells were infected with the supernatant using two centrifugation steps, selected in 800 μg/ml G418 and pools of independent clones used for the analysis.
RESULTS
To investigate the function of the chimeric PAX3/FKHR translocation product in alveolar RMS, we designed a strategy to specifically down-regulate expression of this potential oncogene. For this purpose, RMS cells were incubated with antisense phosphodiester ODN directed against the translational start site of the PAX3 mRNA. Fluorescently labeled ODN were used to optimize their nuclear uptake by absorption to liposomes. The quantity of liposomes used in all subsequent experiments was adjusted to give minimal toxicity vs. maximal ODN uptake. Therefore, cells incubated with liposomes alone were taken as control. For initial experiments, the aRMS cell line Rh30, expressing the PAX3/FKHR fusion protein (11), was used. Cells were incubated with different concentrations of ODN ranging from 0.1–1 μM for 24 hr in serum-free medium. Normal growth medium was added thereafter and viable cells were counted subsequently every day for a total of 5 days. We found that incubation of Rh30 cells with antisense PAX3 ODN resulted in a dose-dependent inhibition of cell growth (Fig. 1a). This effect was not seen with control ODN (sense, missense, or scrambled, see Materials and Methods). Interestingly, a significant drop in cell numbers was observed between day 1 and 2 after ODN addition at 1 μM concentration. Morphological examination revealed that the cells had rounded up and were subsequently floating in the dish, suggesting an actual drop in viability (Fig. 2). Therefore, viability was assessed and expressed in comparison to liposome treatment alone. A significant drop in cell numbers was noticed specifically after PAX3 antisense ODN incubation (Fig. 1b), which was not observed with several control ODN (shown are the results for missense and sense ODN, similar results were obtained with scrambled ODN). As further controls, ODN directed against the PAX7 mRNA that shares close homology to PAX3 (Fig. 1d) did not affect viability. Similarly, survival of unrelated control fibroblast cells not expressing PAX3 was unaffected by treatment with antisense PAX3 ODN (Fig. 1c). Next, activity of the ODN incubations was tested by assessing directly the protein levels of the targeted PAX3/FKHR fusion protein. As shown in Fig. 3a, a significant drop in the level of the fusion protein was observed transiently between day 1 and 2 after treatment with antisense ODN, but not after treatment with missense ODN. The time course of down-regulation of the fusion protein corresponds exactly to the drop of viability observed between day 1 and 2 in the initial experiments. We conclude from these experiments that specific transient down-regulation of the PAX3/FKHR protein in aRMS cells drastically reduces the viability of these cells.
Figure 1.
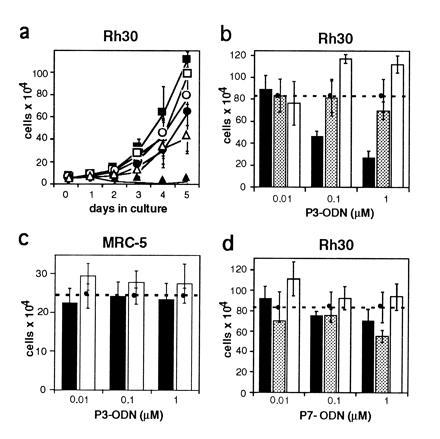
Cell growth and survival rate in the alveolar RMS cell line Rh30 upon treatment with specific PAX3 antisense oligonucleotides. Rh30 cells express the aRMS specific translocation product PAX3/FKHR. ODN are directed against the translational start site of the PAX3 mRNA. (a) Growth rate of Rh30 cells incubated with different concentrations of antisense PAX3 (as-P3, solid symbols) and sense PAX3 (s-P3, open symbols) ODN. Concentrations used were 0.01 μM (▪), 0.1 μM (•), and 1 μM (▴). Viable cells were counted every 24 hr for five consecutive days. (b–d) Viability of Rh30 and control cells incubated with increasing concentrations of antisense (solid bars), missense (shaded bars), or sense (open bars) ODN. The number of cells treated with lipofectin only is indicated by the horizontal lines (dashed lines). Rh30 cells were treated with PAX3 ODN (b) and PAX7 ODN (d), control MRC-5 human lung fibroblasts were treated with PAX3 ODN (c). Cells were counted after 5 days except for in d where they were counted after 3 days. Results shown represent means of at least three independent experiments each using three dishes for every time point counted. Bars = SD.
Figure 2.
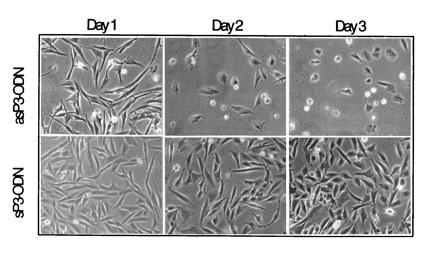
Representative morphology of Rh30 cells treated with either antisense (as-P3) or sense (s-P3) PAX3 ODN after days 1, 2, and 3. Magnification of the phase contrast picture is ×100.
Figure 3.
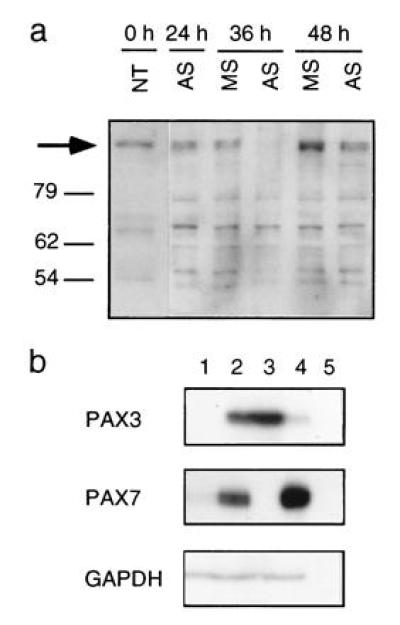
Expression of PAX protein and mRNA. (a) Western blot analysis of Rh30 cell extracts upon treatment with 1 μM antisense (AS) and missense (MS) PAX3 ODN. An equal number of attached cells at day 0, 1, 1.5, and 2 after ODN incubations were loaded on the gel. The arrow indicates the position of the PAX3/FKHR fusion protein. (b) RNase protection assay was performed using 15 μg total RNA isolated from primary human myoblast cells (lane 1), RD (lane 2), Rh30 (lane 3), Rh1 (lane 4), and yeast (lane 5). Details of the probes used for protection analysis have been described previously (13). The use of a probe from the 5′ end does not allow to distinguish between transcripts derived from the wild-type PAX3 or the translocated PAX3/FKHR gene, however the presence of PAX3/FKHR fusion transcripts and protein in Rh30 cells has been described elsewhere (9, 11). Glyceraldehyde-phosphate-dehydrogenase was used as a control for RNA quantities.
The more common subtype of RMS is called embryonal and is characterized by the absence of a specific PAX3/FKHR translocation product. We next investigated the expression levels of wild-type PAX3 and PAX7 mRNA in eRMS cell lines in comparison to primary human myoblasts (Fig. 3b), since both of these proteins are expressed during murine development in muscle precursor cells (6, 16). Using RNase protection experiments, normal human primary myoblasts were found to express low levels of PAX7 mRNA, while no PAX3 message could be detected. In contrast, the eRMS cell line Rh1 (Fig. 3b, lane 4) expressed PAX7 at substantially elevated levels, whereas PAX3 message was barely detectable. A second eRMS cell line (RD, lane 2) expressed PAX7 levels comparable to normal cells, but instead displayed expression of PAX3 mRNA which was not detected in normal myoblasts. Hence, either PAX3 or PAX7 are expressed at elevated levels in these eRMS cells. Consequently, we hypothesized that these wild-type transcription factors could play a similar role in eRMS as found for the PAX3/FKHR fusion protein in aRMS. To test this notion, the two eRMS cell lines RD and Rh1 were treated with ODN targeting either PAX3 or PAX7, as described before. Interestingly, a reduction in viability was observed in both cell lines, but only when treatment was done with the specific antisense ODN against the one PAX message found to be present at elevated levels (Fig. 4 a–d). Rh1 cells, expressing enhanced PAX7 mRNA levels, were affected by treatment with antisense PAX7 ODN only, whereas reduced viability was observed in RD cells treated with antisense PAX3 ODN but not with antisense PAX7 ODN. These experiments further support the observation that treatment of RMS cells with antisense ODN per se (in the absence of the targeted message) has only limited toxicity. Induction of cell death in Rh1 cells was observed already after 24 hr and was slightly more efficient than in RD or Rh30 cells (data not shown). We conclude from these experiments that treatment of eRMS cells with PAX3 or PAX7 antisense ODN allows the induction of cell death, similar to what was observed in aRMS after down-regulation of the PAX3/FKHR fusion protein.
Figure 4.
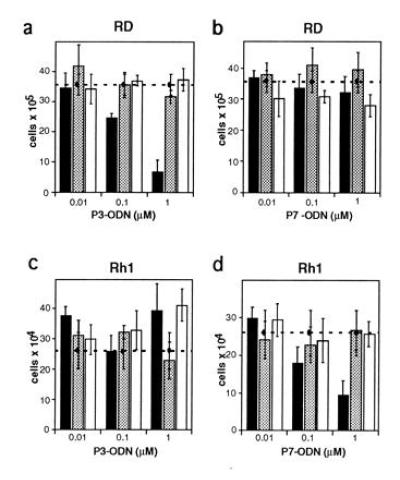
Viability in embryonal RMS cell lines RD (a and b) and Rh1 (c and d) treated with antisense (solid bars), missense (shaded bars), or sense (open bars) oligonucleotides. Viable, attached cells treated with PAX3 (P3, a and c) or PAX7 (P7, b and d) ODN were counted after five days (RD cells) or 3 days (Rh1 cells). The number of cells treated with lipofectin alone is represented by the dashed horizontal line. At least three independent experiments were carried out for each treatment. Bars = SD of the proportion.
The morphological features observed in these experiments, rounding up and membrane blebbing, were similar in all cells analyzed and typical of cells undergoing apoptosis. To distinguish necrosis from apoptosis on the molecular level, two different methods were applied. First, cell cycle analysis was performed after ODN incubations followed by specific DNA staining (Fig. 5 a–f). DNA fragmentation below the G1 peak was evident in all cell lines after specific antisense ODN treatment (Fig. 5 d and f). This fraction of the cells was clearly reduced after control incubations using either sense, missense or scrambled ODN (Fig. 5 a–c, only missense control shown). Second, DNA fragmentation was specifically analyzed in situ using the TUNEL reaction. In these experiments, the percentage of cells displaying DNA fragments was determined 24 hr after ODN treatment (Fig. 5 Right). In Rh1 cells treated with control missense PAX7 ODN, 2.5% (n = 159) of the still attached cells were displaying positive fluorescence. When cells were treated with antisense PAX7 ODN, this fraction increased to 16.9% (n = 112), demonstrating that the rate of apoptosis in the cultures increased specifically after antisense ODN incubation. We conclude from these experiments that apoptosis likely accounts for most of the cell death observed in our antisense experiments.
Figure 5.
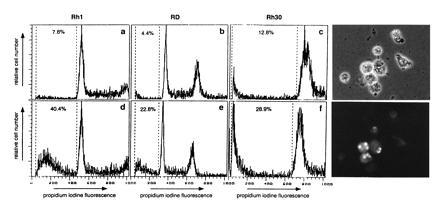
Detection of apoptosis after antisense ODN treatment. (a–f) fluorescence-activated cell sorter cell cycle analysis of the indicated cell lines was performed 60 hr after treatment with 1 μM of either missense ODN (a–c Upper) or antisense ODN (d–f Lower). Attached and floating cells were collected and stained with propidium iodine to reveal DNA content. The percentage of cells below the G1 peak is indicated. (Right) TUNEL labeling of Rh1 cells 24 hr after incubation with 1 μM antisense PAX7 ODN. Phase contrast reveals cells displaying morphological features of apoptosis (Upper), whereas fluorescence microscopy identifies labeled cells (Lower). Quantification of the TUNEL reaction in attached cells reveals that 16.9% (n = 112) of antisense-treated Rh1 cells are labeled vs. 2.5% (n = 159) after sense PAX7 ODN treatment.
The experiments described so far demonstrate that down-regulation of Pax proteins after antisense ODN treatment induces cell death in RMS cells. These observations imply that expression of PAX genes in these embryonal cancer cells is able to suppress apoptosis. To specifically test this notion, we attempted to rescue treatment invoked apoptosis. To this end, Rh1 cells that express PAX7 at high levels and only trace amounts of PAX3 were transduced with a retroviral vector driving the expression of murine Pax3 as well as a control vector expressing β-galactosidase. To control for integration dependent unspecific effects occurring in single clones, pools containing more than 10,000 independent events were formed after selection with G418. First, ectopic expression of Pax3 was assessed using a Pax3 specific antisera. Double immunofluorescence staining revealed varying expression levels with detectable nuclear staining in about 60% of the cells. No staining was observed in the control pool or the parental cells (data not shown). Next, pools of Pax3 as well as control vector transduced cells were treated with antisense and control missense PAX7 ODN (Fig. 6a). The results from three independently conducted experiments demonstrate that the viability in Pax3 transduced cells is significantly improved compared with the control pool. Combining all three experiments, a ratio of 32 ± 8% surviving cells (missense ODN treatment/antisense ODN treatment) in the controls was observed. This ratio increased to 62 ± 18% in Pax3 transduced cells. Since only 60% of the transduced cells displayed detectable ectopic Pax3 expression, complete restoration of cell viability could not be expected. However, when transduced cells were treated with both antisense PAX7 and PAX3 ODN (targeting the ectopically expressed Pax3) the number of surviving cells was again drastically reduced (Fig. 6b). Therefore, ectopic expression of Pax3 in Rh1 cells can, at least partially, protect these cells from apoptosis induced by antisense Pax7 ODN treatment.
Figure 6.
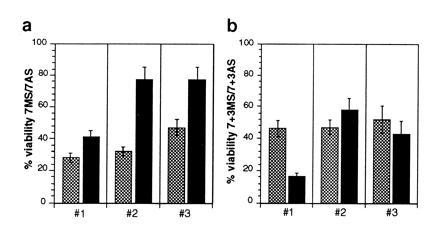
Partial suppression of cell death in Rh1 cells upon retroviral mediated ectopic expression of Pax3. After drug selection, pools of Pax3 (solid bars) and control vector (expressing β-galactosidase; shaded bars) transduced cells were incubated with 1 μM of PAX7 antisense (AS) or missense (MS) ODN and viable cells counted after 48 hr (a). (b) The same cells were treated with both PAX3 and PAX7 antisense or missense ODN at 1 μM each. Viable cells were again counted after 48 hr. For both experiments, results of three independent incubations (1–3) using at least three dishes each for cell number determination are shown expressed as ratio between MS and AS ODN incubations.
DISCUSSION
Here we demonstrate that specific down-regulation of the PAX3/FKHR fusion protein as well as wild-type PAX gene products allows induction of apoptosis in RMS cells. This study provides evidence for an important and novel role of PAX transcription factors in these tumor cells.
We present several lines of evidence supporting the specificity of the antisense approach: (i) unmodified phosphodiester ODN instead of phosphothioate ODN were used; (ii) treatment with control ODN (either sense, missense or scrambled) had only minimal effect on cell viability; (iii) an unspecific activity was reported for a stretch of four guanosine nucleotides in c-myb and c-myc antisense ODN (17). A similar stretch of five guanosine nucleotides was present in our experiments in both antisense and control missense PAX3 ODN, demonstrating that this stretch was not the cause of major unspecificity; (iv) specific transient down-regulation of the PAX3/FKHR protein has been directly demonstrated; (v) there was only minimal toxicity upon treatment of a number of different cells that do not express the targeted PAX gene; and (vi) finally, ectopic expression of Pax3 was able to rescue, at least partially, apoptosis induced by specific antisense PAX7 ODN treatment.
We observed loss of viability both upon down-regulation of the aRMS specific PAX3/FKHR fusion protein as well as wild-type PAX3 and PAX7 in eRMS. These data indicate that these proteins share at least one common function in RMS tumor cells. It has been reported that ectopic expression of several Pax genes in murine embryonal fibroblasts is able to induce transformation and tumor formation in nude mice, suggesting that deregulated Pax proteins can induce tumorigenesis (18). Recently, it was proposed that this effect is, in part, due to loss of expression of the tumor suppressor protein p53 at the transcriptional level (19). In the experiments described in this report, inactivation of p53 by down-regulation of Pax3 and Pax7 does probably not play a major role, mainly for two reasons. (i) Transcriptional repression as a way to inactivate p53 is specific for Pax2 and Pax5 and is not observed by Pax3 (19). (ii) The two cell lines Rh30 and RD analyzed in our study already carry inactivating mutations in p53 (20). Therefore, the observed effects in these two cell lines likely occur independent of p53. How then might these regulators contribute to suppression of the apoptotic pathway? One possibility might be that either activation or repression of specific, so far unknown, target genes is necessary to suppress apoptosis, since Pax3 was demonstrated to be capable of transcriptional activation as well as repression (21). A number of known oncogenes and tumor suppressor genes are involved in the regulation of apoptosis, among them bcl-2 and its family members (22), c-myc (23), pRB (24), and WT1 (25). In some ways, influence on apoptosis by Pax proteins might be comparable, although reversed, to the mechanisms proposed for WT1-induced apoptosis (25). There, the mechanism underlying cell death induction was suggested to involve down-regulation of growth factor signaling. Specifically, down-regulation of the epidermal growth factor receptor by WT1 was demonstrated. Therefore, genes such as growth factors or growth factor receptors might potentially be positively regulated by Pax proteins. On the other hand, an alternative possibility is that specific protein–protein interactions play a major role. Such interactions seem to be important, e.g., control of apoptosis by c-myc (26) and in the bcl-2 protein family (22). In the case of RMS, they would most likely involve the common constituents of PAX3, PAX7, and PAX/FKHR encompassing the two DNA binding domains, namely the paired box and the homeobox. However, in any instances the mechanisms underlying control of apoptosis are still poorly understood and must represent a major focus of research in the future.
Expression of a number of different PAX genes has been correlated with different human tumors. Specifically, PAX2, PAX5, and PAX8 have recently been found at high levels in Wilms tumor (27), medulloblastoma (28), astrocytoma (29), and thyroid tumors (30), respectively. Using a similar antisense approach, growth inhibition was reported upon down-regulation of PAX2 and PAX5 (31, 32). While no loss of viability was reported in B cells upon PAX5 down-regulation, a reduction in cell numbers was observed in renal carcinoma cells upon down-regulation of PAX2 levels (31). This observation raises the possibility that other Pax proteins could also be involved in the regulation of cell death. On the other hand, growth inhibition might also play a role in RMS. Cell cycle analysis demonstrated a decrease in the fraction of cells in G1 upon antisense treatment for Rh1 cells (44.9% to 31.2%; data not shown), whereas we observed an increase in the G1 fraction for RD cells (30.6% to 37.2%; data not shown). Therefore, growth inhibition was not observed in Rh1 cells, in contrast to RD cells where it might occur in addition to induction of apoptosis.
Since it has been suggested that PAX3/FKHR and wild-type PAX3 proteins can inhibit myogenesis after ectopic overexpression in a myogenic mouse cell line, one might expect down-regulation of Pax proteins to result in enhanced differentiation (33). However, we did not observe an increase in multinucleated, differentiated cells as judged by morphological criteria suggesting that in RMS cells additional mechanisms to Pax gene expression are involved in blocking the normal differentiation program.
Suppression of apoptosis by Pax proteins might be part of their complex developmental role. During normal development, Pax3 and Pax7 are expressed in the dermomyotome of the developing somites, whereas only Pax3 is found in the presumptive precursor myoblasts migrating into the limb buds. Interestingly, these cells are absent in homozygous splotch mice lacking functional Pax3 protein, resulting in specific loss of limb muscles (6, 34). Two alternative explanation can be postulated to account for this loss. It might be possible that these cells are not properly recruited to the myogenic lineage and hence the muscle regulatory factors of the basic helix-loop-helix family cannot be activated. On the other hand, our results raise the possibility that lack of survival of the migrating cells might play an additional role in the phenotype of splotch mice.
Nevertheless, these results suggest that new therapeutic avenues can be explored for the treatment of certain malignancies based on inactivation of Pax proteins.
Acknowledgments
We thank O. Murmann for the construction of the pDOL-Pax3 vector and P. Houghton for the generous gift of Rh1 and Rh30 cells. We acknowledge expert technical assistance for the fluorescence-activated cell sorter analysis by Cathrine Felder, and thank Prof. C. W. Heizmann for his continuous support of this project. The work is supported by the Swiss National Science Foundation Grant 3100–37378.93.
Footnotes
Abbreviations: RMS, rhabdomyosarcoma; FKHR, forkhead domain; ODN, oligonucleotide; aRMS, alveolar RMS; eRMS, embryonal RMS; TUNEL, terminal transferase-mediated dUTP nick-end labeling.
References
- 1.Stuart E T, Gruss P. Hum Mol Genet. 1995;4:1717–1720. doi: 10.1093/hmg/4.suppl_1.1717. [DOI] [PubMed] [Google Scholar]
- 2.Strachan T, Read A P. Curr Opin Genet Dev. 1994;4:427–438. doi: 10.1016/0959-437x(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 3.Torres M, Gomezpardo E, Dressler G R, Gruss P. Development (Cambridge, UK) 1995;121:4057–4065. doi: 10.1242/dev.121.12.4057. [DOI] [PubMed] [Google Scholar]
- 4.Urbanek P, Wang Z, Fetka I, Wagner E F, Busslinger M. Cell. 1994;79:901–912. doi: 10.1016/0092-8674(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 5.Hill R, Van Heiningen V. Trends Genet. 1992;8:119–120. doi: 10.1016/0168-9525(92)90359-C. [DOI] [PubMed] [Google Scholar]
- 6.Bober E, Franz T, Arnold H H, Gruss P, Tremblay P. Development (Cambridge, UK) 1994;120:603–612. doi: 10.1242/dev.120.3.603. [DOI] [PubMed] [Google Scholar]
- 7.Williams B A, Ordahl C P. Development (Cambridge, UK) 1994;120:785–796. doi: 10.1242/dev.120.4.785. [DOI] [PubMed] [Google Scholar]
- 8.Galili N, Davis R J, Fredericks W J, Mukhopadhyay S, Rauscher F J, Emanuel B S, Rovera G, Barr F G. Nat Genet. 1993;5:230–235. doi: 10.1038/ng1193-230. [DOI] [PubMed] [Google Scholar]
- 9.Shapiro D N, Sublett J E, Li B T, Downing J R, Naeve C W. Cancer Res. 1993;53:5108–5112. [PubMed] [Google Scholar]
- 10.Davis R J, D’Cruz C M, Lovell M A, Biegel J A, Barr F G. Cancer Res. 1994;54:2869–2872. [PubMed] [Google Scholar]
- 11.Fredericks W J, Galili N, Mukhopadhyay S, Rovera G, Bennicelli J, Barr F G, Rauscher F J. Mol Cell Biol. 1995;15:1522–1535. doi: 10.1128/mcb.15.3.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sublett J E, Jeon I S, Shapiro D N. Oncogene. 1995;11:545–552. [PubMed] [Google Scholar]
- 13.Schäfer B W, Czerny T, Bernasconi M, Genini M, Busslinger M. Nucleic Acids Res. 1994;22:4574–4582. doi: 10.1093/nar/22.22.4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Korman A J, Frantz J D, Strominger J L, Mulligan R C. Biochemistry. 1987;84:2150–2154. doi: 10.1073/pnas.84.8.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Price J, Turner D, Cepko C. Proc Natl Acad Sci USA. 1987;84:156–160. doi: 10.1073/pnas.84.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jostes B, Walther C, Gruss P. Mech Dev. 1991;33:27–38. doi: 10.1016/0925-4773(90)90132-6. [DOI] [PubMed] [Google Scholar]
- 17.Burgess T L, Fisher E F, Ross S L, Bready J V, Qian Y X, Bayewitch L A, Cohen A M, Herrera C J, Hu S S, Kramer T B, Lott F D, Martin F H, Pierce G F, Simonet L, Farrel C L. Proc Natl Acad Sci USA. 1995;92:4051–4055. doi: 10.1073/pnas.92.9.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maulbecker C C, Gruss P. EMBO J. 1993;12:2361–2367. doi: 10.1002/j.1460-2075.1993.tb05890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stuart E T, Haffner R, Oren M, Gruss P. EMBO J. 1995;14:5638–5645. doi: 10.1002/j.1460-2075.1995.tb00251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Felix C A, Kappel C C, Mitsudomi T, Nau M M, Tsokos M, Crouch G D, Nisen P D, Winick N J, Helman L J. Cancer Res. 1992;52:2243–2247. [PubMed] [Google Scholar]
- 21.Chalepakis G, Jones F S, Edelman G M, Gruss P. Proc Natl Acad Sci USA. 1994;91:12745–12749. doi: 10.1073/pnas.91.26.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hockenbery D M. J Cell Sci. 1994;55:51–55. doi: 10.1242/jcs.1994.supplement_18.7. [DOI] [PubMed] [Google Scholar]
- 23.Evan G I, Brown L, Whyte M, Harrington E. Curr Opin Cell Biol. 1995;7:825–834. doi: 10.1016/0955-0674(95)80066-2. [DOI] [PubMed] [Google Scholar]
- 24.Cordon-Cardo C. Am J Pathol. 1995;147:545–560. [PMC free article] [PubMed] [Google Scholar]
- 25.Englert C, Hou X, Maheswaran S, Bennett P, Ngwu C, Re G G, Garvin A J, Rosner M R, Haber D A. EMBO J. 1995;14:4662–4675. doi: 10.1002/j.1460-2075.1995.tb00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kohlhuber F, Hermeking H, Graessmann A, Eick D. J Biol Chem. 1995;270:28797–28805. doi: 10.1074/jbc.270.48.28797. [DOI] [PubMed] [Google Scholar]
- 27.Dressler G R, Douglass E C. Proc Natl Acad Sci USA. 1992;89:1179–1183. doi: 10.1073/pnas.89.4.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kozmik Z, Sure U, Ruedi D, Busslinger M, Aguzzi A. Proc Natl Acad Sci USA. 1995;92:5709–5713. doi: 10.1073/pnas.92.12.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stuart E T, Kioussi C, Aguzzi A, Gruss P. Clin Cancer Res. 1995;1:207–214. [PubMed] [Google Scholar]
- 30.Fabbro D, Diloreto C, Beltrami C A, Belfiore A, Dilauro R, Damante G. Cancer Res. 1994;54:4744–4749. [PubMed] [Google Scholar]
- 31.Gnarra J R, Dressler G R. Cancer Res. 1995;55:4092–4098. [PubMed] [Google Scholar]
- 32.Wakatsuki Y, Neurath M F, Max E E, Strober W. J Exp Med. 1994;179:1099–1108. doi: 10.1084/jem.179.4.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Epstein J A, Lam P, Jepeal L, Maas R L, Shapiro D N. J Biol Chem. 1995;270:11719–11722. doi: 10.1074/jbc.270.20.11719. [DOI] [PubMed] [Google Scholar]
- 34.Goulding M, Lumsden A, Paquette A J. Development (Cambridge, UK) 1994;120:957–971. doi: 10.1242/dev.120.4.957. [DOI] [PubMed] [Google Scholar]