

Abstract
Cerebral deposition of the amyloid β protein (Aβ) is an early and invariant feature of Alzheimer disease (AD). Whereas the 40-amino acid form of Aβ (Aβ40) accounts for ≈90% of all Aβ normally released from cells, it appears to contribute only to later phases of the pathology. In contrast, the longer more amyloidogenic 42-residue form (Aβ42), accounting for only ≈10% of secreted Aβ, is deposited in the earliest phase of AD and remains the major constituent of most amyloid plaques throughout the disease. Moreover, its levels have been shown to be increased in all known forms of early-onset familial AD. Thus, inhibition of Aβ42 production is a prime therapeutic goal. The same protease, γ-secretase, is assumed to generate the C termini of both Aβ40 and Aβ42. Herein, we analyze the effect of the compound MDL 28170, previously suggested to inhibit γ-secretase, on β-amyloid precursor protein processing. By immunoprecipitating conditioned medium of different cell lines with various Aβ40- and Aβ42-specific antibodies, we demonstrate a much stronger inhibition of the γ-secretase cleavage at residue 40 than of that at residue 42. These data suggest that different proteases generate the Aβ40 and Aβ42 C termini. Further, they raise the possibility of identifying compounds that do not interfere with general β-amyloid precursor protein metabolism, including Aβ40 production, but specifically block the generation of the pathogenic Aβ42 peptide.
The 39- to 43-amino acid amyloid β protein (Aβ) is deposited as amyloid in the brains of all patients with Alzheimer disease (AD). It has been shown that the primary component of deposits in the cerebral vasculature is short Aβ, ending with a C terminus at residue 39 or 40 (Aβ40) (1, 2), whereas long Aβ, ending with a C terminus at residue 42 (Aβ42), accumulates initially and predominantly in parenchymal plaques (3–7). Aβ is constitutively secreted by a wide variety of cells and exists in a soluble form in biological fluids (8, 9). During the last 2 years, evidence has accumulated suggesting that Aβ42 plays the key role in the process of plaque formation. (i) In vitro data demonstrate that Aβ42 accelerates the formation of Aβ fibrils by a nucleation dependent mechanism (10). (ii) While accounting for only 10% of total Aβ secreted from cells (roughly 90% is Aβ40) (11, 12), Aβ42 is the major plaque component (3, 4). Furthermore, all three early-onset familial AD genes identified to date have been shown to lead to an increase in Aβ42. Only the Swedish β-amyloid precursor protein (βAPP) missense mutation also increases Aβ40 (11), whereas the βAPP717 mutations and the recently described presenilin 1 and 2 mutations do not (13, 14). Thus, the long Aβ42 peptide appears to be a prime target for therapeutic intervention. However, none of the enzymes involved in the major steps of βAPP processing have been identified, including γ-secretase, the protease that generates the C terminus of Aβ. It has generally been assumed that the same enzyme(s) generate both Aβ40 and Aβ42 and it has been shown that both peptides share a common secretory mechanism that involves acidic compartments such as the late Golgi or early endosomes (12). Recently, Higaki et al. (15) have shown that the calpain inhibitor MDL 28170 inhibits the production of both total Aβ and total p3 and leads to an accumulation of their respective 12-kDa and 10-kDa βAPP precursor fragments in cells, suggesting a direct inhibition of γ-secretase. Using end specific antibodies, we show herein that this compound primarily decreases Aβ40 and p340 whereas Aβ42 and p342 are only marginally affected, indicating that pharmacologically dissociable pathways and potentially different γ-secretases are responsible for cleavage at residue 40 and at residue 42 of Aβ. The therapeutic implications of this surprising result are discussed.
MATERIALS AND METHODS
Cell Lines.
All transfected cell lines described in this paper carry derivatives of pCMV695, a plasmid carrying βAPP695 under control of the cytomegalovirus (CMV) promoter (16). K695sw are human embryonic kidney 293 cells stably transfected with a construct carrying the AD-linked double (“Swedish”) mutation K595N/M596L (17); K695717I are 293 cells stably transfected with βAPP695 carrying the mutation F717I (APP770 numbering). CHO695 are Chinese hamster ovary cells stably transfected with pCMV695. SKN695 are SK-N-SH human neuroblastoma cells stably transfected with pCMV695.
Pulse–Chase Experiments and Immunoprecipitations.
To analyze the effect of MDL 28170 on the processing of βAPP, cells were grown to confluence in two 10-cm dishes, pulse-labeled with 600 μCi (1 Ci = 37 GBq) of [35S]methionine in 4 ml of serum-free medium for 2 h and then chased for 2 h with 4 ml of medium containing 10% fetal bovine serum and the indicated final concentration of MDL 28170 [initially dissolved at 200 mM in dimethyl sulfoxide (DMSO)]. Control dishes were treated with DMSO alone. Conditioned media and cell lysates were analyzed by immunoprecipitation, as described (18). Polyclonal antibody R1736 to residues 595–611 of APP695 was used to precipitate α-APPs (19). This antibody recognizes an epitope that is specific for the free C terminus of α-cleaved APPs. Polyclonal antibody R1282 was generated to synthetic Aβ1–40. This antibody precipitates total Aβ and p3 (and small variable amounts of APPs) from the medium of cultured cells (18). Polyclonal antibody 1963 was raised to synthetic Aβ21–37 (18). The monoclonal antibody 2G3, specific for Aβ x-40, was produced by injecting female A/J mice intraperitoneally with 100 μg of immunogen per injection. The immunogen consisted of the peptide NH2-CYS-NH-CH2-(CH2)5-CO-GLMVGGVV-COOH coupled to sheep anti-mouse IgG by using maleimidohexanoyl-N-hydroxysuccinimide. The immunogen was emulsified with Freund’s complete adjuvant for the first immunization, and all subsequent immunizations were with 100 μg of immunogen emulsified with Freund’s incomplete adjuvant at approximately 2-week intervals. Three days before fusion, a mouse was injected with two 50-μg quantities of immunogen, one intravenously and one intraperitoneally. Three days after injection, the spleen was removed, and splenocytes were isolated and fused with SP2/0 mouse myeloma by a modification of published methods (20). The resulting hybridoma cells were screened for antibody with the ability to capture 125I-labeled Aβ1–40 in solution by immunoprecipitation. Antibody 2G3 reacts strongly with Aβ1–40 but has essentially no cross-reactivity with Aβ1–42. Twenty micrograms of this antibody was used to immunoprecipitate the chase medium of two dishes. The monoclonal antibody 21F12 was produced as described for 2G3 using as immunogen the synthetic peptide NH2-CYS-NH-CH2-(CH2)5-CO-GLMVGGVVIA-COOH. This antibody has very high specificity for Aβ1–42 over Aβ1–40. At a concentration of 20 ng/ml of both peptides, less than 0.4% cross-reactivity was observed. Twenty micrograms of this antibody was used to immunoprecipitate Aβ42 and p342 from the chase medium of two dishes. The monoclonal antibody BCO5 specifically detects Aβ42 and p342 (13, 14). One hundred micrograms of this antibody was used to immunoprecipitate the chase medium of three dishes. The polyclonal antibody C42 specifically detects Aβ42 and p342 (21). Serum was used at a 1:50 dilution to immunoprecipitate the chase medium of three dishes. The polyclonal antibody C7 against the last 20 residues of the βAPP cytoplasmic tail (22) precipitates N′- and N′- plus O′-glycosylated full-length βAPP as well as its C-terminal proteolytic fragments. The antibody sw192 (23, 24) specifically precipitates β-cleaved APPs carrying the Swedish mutation. SDS/PAGE of immunoprecipitates of cell extracts or of Aβ from medium was carried out on 10–20% Tris/Tricine gels (NOVEX, San Diego), whereas APPs precipitates were electrophoresed on 10% SDS/polyacrylamide Tris/glycine gels. All quantitations were performed with a PhosphorImager model 400A using Image-QuaNT software (Molecular Dynamics).
RESULTS
Inhibition of βAPP Processing by the MDL 28170 Inhibitor.
We first set out to reproduce the results of Higaki et al. (15) on the action of MDL 28170 on βAPP processing using human kidney 293 cells stably expressing βAPP695 with the Swedish familial AD mutation (K695sw). These experiments were done using a pulse–chase paradigm: K695sw cells were labeled for 2 h and then chased for 2 h in the presence or absence of 200 μM MDL 28170. Aliquots of the chase media from treated and untreated cells were subjected to SDS/PAGE. No decrease in the amounts of the major secreted cellular proteins was detected; only an increase in some low molecular weight proteins was observed (Fig. 1, lanes 1 and 2), suggesting that under the conditions of the experiment, MDL 28170 does not interfere with general protein secretion. We next analyzed the chase medium for changes in the amounts of α- and β-cleaved APPs by using antibodies specific for each form. Antibody 1736 specifically immunoprecipitates α-cleaved APPs (19, 24). This antibody revealed an increase in α-APPs production upon treatment (Fig. 1, lanes 3 and 4), indicating that MDL 28170 does not inhibit but rather stimulates α-secretase in kidney cells. However, no stimulation was observed in CHO cells (data not shown and ref. 15). 192sw specifically immunoprecipitates the β-cleaved APPs species ending with the Swedish mutant Leu596 (23). Immunoprecipitation with this antibody did not reveal a significant change in β-secretase activity (Fig. 1, lanes 5 and 6).
Figure 1.
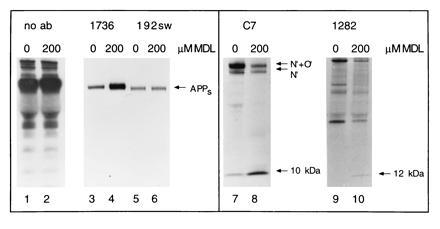
MDL 28170 influences βAPP metabolism. 0, Untreated K695sw cells. 200, K695sw cells treated with 200 μM MDL 28170. Lanes: 1 and 2, aliquot of total chase medium electrophoresed directly on the gel; 3 and 4, 1736 immunoprecipitation of chase medium; 5 and 6, 192sw immunoprecipitation of chase medium [note that β-cut APPs (lanes 5 and 6) runs slightly below α-cut APPs (lanes 3 and 4), as expected]; 7 and 8, C7 immunoprecipitation of cell lysates (N′ and N′/O′-glycosylated forms of full-length βAPP as well as the 10-kDa C-terminal fragment are indicated); 9 and 10, 1282 immunoprecipitation of cell lysates.
We next analyzed cell lysates for changes in full-length βAPP and its C-terminal fragments by using antibody C7, directed to the last 20 amino acids of βAPP (22). This antibody precipitates N′- and N′/O′-glycosylated full-length βAPP and its 10-kDa C-terminal fragment that remains membrane bound after α-secretase cleavage. Upon treatment with MDL 28170, a striking increase in the level of the 10-kDa C-terminal fragment was observed, but the decrease in full-length βAPP was not consistently seen (Fig. 1, lanes 7 and 8). In K695sw cells, the 12-kDa C-terminal fragment which remains membrane bound after β-secretase cleavage cannot be easily resolved and detected by antibody C7 (25). We therefore tried to precipitate this fragment using antibody 1282 raised to synthetic Aβ1–40. This antibody, whose dominant epitope is within the first 16 residues of Aβ (i.e., N-terminal to the α-secretase cleavage site), can precipitate the 12-kDa but not the 10-kDa fragment, and therefore, the faint 12-kDa band is not overshadowed by the much more abundant 10-kDa band (26). Whereas no 12-kDa fragment was detectable in untreated cells, this band was clearly observed upon treatment with the inhibitor (Fig. 1, lanes 9 and 10). In summary, no inhibition of α- or β-secretase cleavage but, rather, an increase in both 10- and 12-kDa C-terminal fragments was observed upon treatment with MDL 28170, strongly supporting the postulated role of this compound as inhibiting γ-secretase either directly or indirectly.
MDL 28170 Inhibits the Production of Aβ40 and p340 but not Aβ42 and p342.
MDL 28170 had previously been shown to inhibit the secretion of both Aβ and p3 and had, therefore, been suggested to lead to inhibition of γ-secretase. This inhibition was observed by immunoprecipitating medium from treated cells with a polyclonal antibody raised to synthetic Aβ1–40 (15). Because the vast majority of secreted Aβ and p3 peptides end at amino acid 40, this experiment does not distinguish whether only the major γ-secretase cleavage at position 40 or also the less frequent γ-secretase cleavage at position 42 is inhibited. To address this question, we performed pulse–chase experiments on K695sw cells using different doses of MDL 28170, followed by sequential immunoprecipitation of the same medium first with 21F12 (a monoclonal antibody that specifically precipitates Aβ peptides ending at position 42) and then with 1282 (that precipitates all forms of Aβ and p3) (Fig. 2A). Interestingly, 21F12 precipitated not only Aβ but also p3 peptides, thus, demonstrating the existence of secreted p342, which had not been described before. Total Aβ and total p3 were strongly and significantly decreased with doses of MDL 28170 greater than 50 μM (e.g., at 200 μM, P < 0.001). In contrast, Aβ42 and p342 showed a bell-shaped dose–response curve, with only a small and insignificant decrease at 200 μM, the dose used by Higaki et al. (15) and the highest dose tested herein. Using MDL 28170 at 200 μM, the experiment was repeated four times and the results were quantitated by phosphorimaging (Fig. 2B). These data indicate that, under the conditions described above, the differential effect of the inhibitor is significant for both the Aβ and the p3 peptides.
Figure 2.
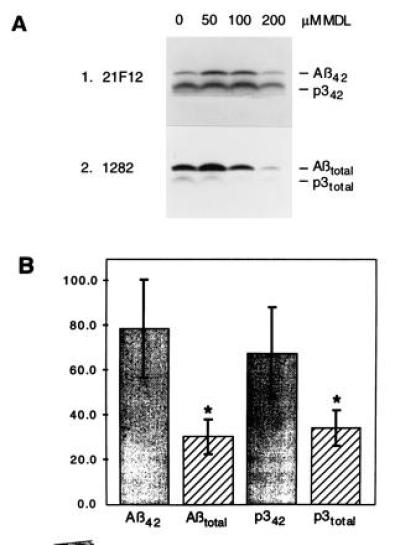
Differential inhibition of Aβ42 and Aβ40 formation. (A) Labeled K695sw cells were chased with the indicated concentrations of MDL 28170 and precipitated with 21F12 (Upper) followed by 1282 (Lower). (B) Quantitation of the effect of 200 μM MDL 28170 on Aβ and p3 by phosphorimaging. The bars show the pixel number relative to an untreated control (200); SDs are indicated. The decreases in Aβtotal and p3total relative to an untreated control were significant (∗, two-tailed t test, n = 4, P < 0.001) and the decreases in Aβ42 and p342 upon treatment with MDL 28170 did not reach significance. Moreover, the difference in inhibition of Aβ42 versus total Aβ is significant (two-tailed t test, n = 4, P < 0.01) and the difference in inhibition of p342 versus total p3 is also significant (two-tailed t test, n = 4, P < 0.05).
Differential Inhibition Is Accomplished Under a Number of Conditions.
To confirm that the differential effect observed in the Aβ40/Aβ42 and p340/p342 precipitations is meaningful, we performed a number of control experiments using the K695sw cells. First, we treated K695sw cells in a 2-h pulse/2-h chase paradigm with 1 μM of the phorbol ester, phorbol 12,13-dibutyrate, which has been shown to decrease total Aβ but increase total p3, probably by diverting βAPP substrate from the β-secretase to the α-secretase proteolytic pathway (27, 28). This effect should be independent of the subsequent γ-secretase cleavage, and thus the 40- and 42-amino acid forms of each metabolite should be equally decreased or increased if the immunoprecipitation paradigm used herein works correctly. Indeed, when conditioned medium of phorbol 12,13-dibutyrate-treated cells was precipitated with 21F12, the expected decrease in Aβ42 and increase in p342 were observed (Fig. 3A), indicating that the Aβ42 immunoprecipitation signal does reflect changes in the amounts of precipitable material. Subsequent immunoprecipitation with R1282 showed the same expected effects for total Aβ and total p3 (Fig. 3A).
Figure 3.
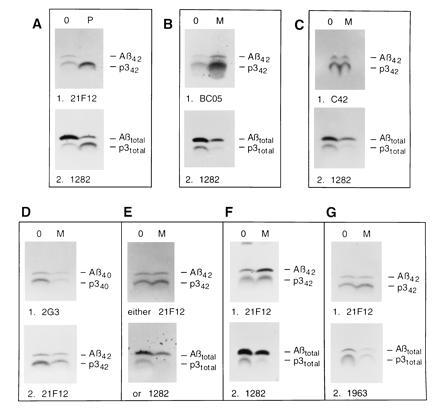
Differential inhibition of Aβ42 and Aβ40 formation in K695sw cells under a variety of conditions. (A) Treatment with 1 μM phorbol 12,13-dibutyrate (lanes P) decreases both Aβ42 and Aβtotal. (Upper) 21F12 precipitation. (Lower) Subsequent 1282 precipitation. (B–E) Cells were chased with (lanes M) or without (lanes 0) 200 μM MDL 28170 and the resultant media were immunoprecipitated as follows. (B) First with BC05 (Upper) and then with 1282 (Lower). (C) First with C42 (Upper) and then with 1282 (Lower). (D) First with 2G3 (Upper) and then with 21F12 (Lower). (E) In two separate aliquots, either with 21F12 (Upper) or with 1282 (Lower). (F) Cells were labeled for 3 h in the presence of 100 μM MDL 28170 and the medium was immunoprecipitated first with 21F12 (Upper) and then with 1282 (Lower). (G) Cells were chased with (lane M) or without (lane 0) 200 μM MDL 28170 and the resultant media were immunoprecipitated first with 21F12 (Upper) and then with 1963 (Lower).
The conclusion that Aβ42 and p342 are not decreased by MDL 28170 depends critically on the specificity of the 21F12 antibody. To confirm the effects observed with this antibody, two previously well-characterized Aβ42-end-specific antibodies were used in the same pulse–chase paradigm with MDL 28170 at 200 μM. The monoclonal antibody BC05 has been extensively used in ELISA assays to detect Aβ42 (12–14, 29), When medium from K695sw cells treated with MDL 28170 was precipitated with this antibody, we observed an actual increase in both Aβ42 and p342. The subsequent precipitation with R1282 showed the usual decrease in total Aβ and p3 (Fig. 3B). The polyclonal antibody C42 has also been shown to be specific for Aβ42 (21). Likewise, this antibody did not show a decrease in Aβ42 and p342 upon MDL 28170 treatment, whereas the subsequent precipitation with R1282 showed the usual decrease in total Aβ and p3 (Fig. 3C). The decrease in Aβ40 and p340 was also found when 2G3, a monoclonal antibody specific for the free C terminus of Aβ40 and p340 was used to precipitate first, followed by precipitation with 21F12 (Fig. 3D). The differential inhibition of Aβ production was also detected when the precipitations of the chase medium were carried out not sequentially, as described above, but in parallel. That is, aliquots of media from treated and untreated cells were precipitated with 21F12 to detect the Aβ42 forms, and other aliquots were precipitated with 1282 to detect total Aβ and total p3. This parallel precipitation produced the same result as the sequential precipitations described above (Fig. 3E). In summary, three different Aβ42-end-specific antibodies consistently show that MDL 28170 does not significantly decrease Aβ42 and p342 production, whereas a monoclonal antibody to Aβ40 and p340 and different polyclonal antibodies to total Aβ and p3 demonstrate strong decreases in these peptides. In addition, immunoprecipitation with 21F12 followed by 1282 again revealed this differential inhibition when the inhibitor (100 μM) was applied during a 3-h labeling period instead of in the chase phase of a pulse–chase experiment (Fig. 3F). Finally, the decrease in total Aβ and total p3 was also detected when 1282 was replaced by 1963, a polyclonal antibody raised to Aβ21–37 (18) (Fig. 3G).
Differential Inhibition Is Observed in Several Cell Lines.
To determine whether the differential inhibition is specific for K695sw, three additional cell lines were treated in the standard 2-h pulse/2-h chase paradigm, and the conditioned medium was precipitated first with 21F12 and then with 1282. The kidney cell line K695717I expresses APP695 carrying the 717I mutation. This line was chosen because the mutation results in increased production of Aβ42 (13). At 200 μM MDL 28170, no decrease of Aβ42 and p342 was observed, whereas Aβ40 and p340 were strongly reduced (Fig. 4A). The CHO cell line CHO695 stably transfected with wild-type βAPP695 cDNA was treated with 200 μM MDL 28170, and only a slight decrease of Aβ42 and p342 was observed, whereas Aβ40 and p340 were markedly reduced (Fig. 4B). The human neuroblastoma cell-line SKN695, expressing wild-type βAPP695 was treated with 200 μM MDL 28170 (Fig. 4C). While Aβ42 and p342 were slightly increased, total Aβ and total p3 were substantially decreased. Thus, differential inhibition of Aβ42 versus Aβ40 and p342 versus p340 production is not only observed in K695sw but also in a cell line with an AD-linked βAPP717 missense mutation, in a hamster cell line and in a human neural cell line expressing wild-type βAPP.
Figure 4.
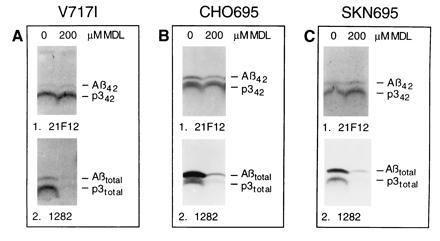
Differential inhibition of Aβ42 and Aβ40 formation in different cell types. Labeled cells were chased with the indicated concentrations of MDL 28170, and the media were precipitated with 21F12 (Upper) followed by 1282 (Lower). (A) K695717I cells. The relatively low βAPP expression in this line leads to a faint Aβ42 band. However, the bands are not due to endogenous βAPP, because nontransfected 293 cells do not show any Aβ or p3 bands under the conditions of the experiment (not shown). (B) CHO695 cells. Note that in CHO cells, the p3 bands migrate as doublets, as described (30). (C) SKN695 cells.
DISCUSSION
Several lines of evidence strongly support the hypothesis that progressive cerebral deposition of Aβ is a seminal event in the pathogenesis of AD (for review, see ref. 31). As a result, there is considerable interest in developing pharmacological inhibitors of the proteases responsible for constitutively generating Aβ from βAPP, i.e. the so-called β- and γ-secretases. Although several known enzymes have been proposed as candidates for these proteases, definitive identification of the responsible enzymes has not yet been reported. Recent investigations on the C-terminal microheterogeneity of Aβ have very strongly suggested that the longer Aβ42 form, although accounting for only for a minor portion of total secreted Aβ, plays the key role in plaque formation (3, 4, 29) and is increased in all forms of early-onset familial AD that have been analyzed (13, 14). These findings make the specific reduction of Aβ42 load a prime therapeutic goal. To address this problem, two questions must be answered. (i) Are Aβ40 and Aβ42 generated in two fundamentally different pathways? (ii) If this is not the case, does the same γ-secretase cleave βAPP molecules to generate both Aβ40 and Aβ42 or are different enzymes with different specificities involved?
Because most previous mechanistic and pharmacological work has measured total Aβ, the influence of various compounds and AD-linked βAPP mutations on Aβ42 production is unknown. However, the available data suggest that Aβ40 and Aβ42 are not generated in entirely different pathways. First, it was shown that the Swedish βAPP mutation, which increases β-secretase cleavage, leads to the same relative increase of Aβ40 and Aβ42 (11), suggesting that both Aβ forms share the same β-secretase cleavage. Then, the agents monensin, brefeldin A, NH4Cl, and bafilomycin A1 were shown to inhibit both Aβ40 and Aβ42 in a parallel and concentration-dependent manner (12). All of these agents had previously been shown to inhibit the production of total Aβ by acting on protein trafficking or β-secretase (23, 32, 33). Finally, we show herein that phorbol ester inhibits both Aβ42 and total Aβ production (Fig. 3). In summary, these data suggest that the initial steps of Aβ40 and Aβ42 generation occur in the same pathway, perhaps up to the point of γ-secretase cleavage. Thus, the same 12-kDa precursor can give rise to both Aβ40 and Aβ42. By analogy, one would postulate that the same 10-kDa precursor can be the substrate for both p340 and p342 production. Indeed, we clearly demonstrate herein the existence of secreted p342 by immunoprecipitation and show that its amount rises in parallel with the amount of total p3 upon treatment with phorbol ester (Fig. 3).
The generation of Aβ40 and Aβ42 from the same 12-kDa precursor could be accomplished if the same γ-secretase enzyme always cleaves 90% between residues 40 and 41 and 10% between residues 42 and 43. Alternatively, one could postulate that the same protease generates both Aβ40 and Aβ42 but that the ratio of the two peptides depends on the localization of the enzyme to a specific vesicle and the intravesicular conditions (e.g., pH). Finally, one could invoke two different proteases with different specificities and potentially different inhibition profiles.
To address this mechanistically and therapeutically important question, we have analyzed the only published compound that leads to inhibition of γ-secretase, based on its effect on total Aβ and total p3 (15). We have reproduced the findings of Higaki et al. (15) on this compound, MDL 28170, for total Aβ and other βAPP metabolites in our cell lines. However, we document a striking differential effect at 200 μM: a much stronger inhibition of cleavage at residue 40 than at residue 42 for both Aβ and p3 in human kidney cells stably transfected with APP695 carrying the Swedish mutation. This significant difference was found by immunoprecipitating first with a monoclonal Aβ42-end-specific antibody and then reprecipitating with a polyclonal antibody for total Aβ and p3. The same result was obtained when the precipitations were carried out in parallel rather than sequentially. The effect could also be demonstrated using a monoclonal Aβ40-end-specific antibody instead of the polyclonal Aβ antibody for total Aβ. Our initial Aβ42-specific immunoprecipitations showed a high ratio of p3 to Aβ (Fig. 2). However, the Aβ40-specific antibody also produced a high p3/Aβ ratio, excluding the possibility that the 10-kDa precursor of p3 is more susceptible to cleavage at residue 42 than is the 12-kDa precursor of Aβ and suggesting instead that, these end-specific antibodies precipitate p3 more avidly than general Aβ antisera like 1282, which contains some antibodies to epitopes N-terminal to p3. Most importantly, the differential inhibition of residue 40 versus 42 cleavage could also be demonstrated when the extensively characterized Aβ42-specific antibody BC05 (12–14, 29) was used (Fig. 3). Three additional distinct cell lines, including a human neuroblastoma line showed the same effect (Fig. 4), demonstrating the generality of the phenomenon.
We conclude that two distinct activities cleave at residues 40 and 42. These activities could be performed by the same enzyme and MDL 28170 would then act indirectly to downregulate the residue 40 cleavage, e.g., by changing intravesicular pH. However, all previously tested agents that change pH, such as NH4Cl and bafilomycin A1, and compounds that block trafficking inhibit both Aβ40 and Aβ42 in parallel (12). We therefore favor the alternative hypothesis that different enzymes with different inhibition profiles cleave at residues 40 and 42. Future studies should address whether there are subtle differences in the kinetics of residue 40 versus 42 cleavage or in the subcellular localization of the different γ-secretase activities. Our data demonstrating a preferential inhibition of residue 40 cleavage by the compound MDL 28170 raise the important issue that pharmaceutical screens for inhibition of total Aβ and p3 production should be followed by a residue 42-specific assay to analyze whether only the production of Aβ40 and p340 is substantially inhibited. However, the principal conclusion arising from the data presented herein is that they suggest the possibility of identifying compounds that selectively inhibit the residue 42 cleavage without affecting 90% of total Aβ production and most of APP processing. We propose that this may be a more promising strategy to decrease plaque amyloid burden in AD than the previously discussed approach to attempt the total inhibition of γ-secretase cleavage. The latter strategy requires the cells to metabolize the high amounts of accumulated 10- and 12-kDa fragments for the entire time of inhibitor treatment, but these fragments have been demonstrated to be potentially neurotoxic, e.g., see refs. 34–36. In contrast, specific inhibition of Aβ42 production would increase the load of 10-kDa and 12-kDa fragments only slightly, or perhaps not at all if they actually undergo compensatory cleavages by the residue 40-specific γ-secretase. A partial pharmacological inhibition of Aβ42 production to reach near normal Aβ42 levels in the brain could probably prevent or delay the onset of AD in carriers of the early onset APP717 mutations (13) and perhaps also in carriers of the presenilin mutations (14), because both of these aggressive forms of familial AD involve a highly selective elevation of Aβ42 production. Of related interest is the evidence that very young subjects with Down syndrome selectively develop Aβ42-specific plaques as early as age 12, many years before substantial Aβ40 deposition occurs (37).
Acknowledgments
We thank Dr. S. Younkin for the antibody BC05, Dr. T. C. Saido for the antibody C42, and Dr. B. Cordell and Hoechst Marion Roussel, Inc., for the generous gift of MDL 28170. This work was supported in part by National Institutes of Health Grants AG05134 and AG12749 to D.J.S.
Footnotes
Abbreviations: Aβ, amyloid β protein; AD, Alzheimer disease; βAPP, β-amyloid precursor protein.
References
- 1.Joachim C L, Duffy L K, Morris J H, Selkoe D J. Brain Res. 1988;474:100–111. doi: 10.1016/0006-8993(88)90673-7. [DOI] [PubMed] [Google Scholar]
- 2.Castano E M, Frangione B. Lab Invest. 1988;58:122–132. [PubMed] [Google Scholar]
- 3.Roher A E, Lowenson J D, Clarke S, Woods A S, Cotter R J, Gowing E, Ball M J. Proc Natl Acad Sci USA. 1993;90:10836–10840. doi: 10.1073/pnas.90.22.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina H, Ihara Y. Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 5.Iwatsubo T, Mann D M, Odaka A, Suzuki N, Ihara Y. Ann Neurol. 1995;37:294–299. doi: 10.1002/ana.410370305. [DOI] [PubMed] [Google Scholar]
- 6.Yamaguchi H, Sugiahra S, Ishiguro K, Takashima A, Hirai S. Amyloid Int J Clin Invest. 1995;2:7–16. [Google Scholar]
- 7.Mann D M A, Iwatsubo T, Ihara Y, Cairns N J, Lantos P L, Bogdanovic N, Lannfelt L, Winblad B, Maatschieman M L C, Rossor M N. Am J Pathol. 1996;148:1257–1266. [PMC free article] [PubMed] [Google Scholar]
- 8.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M G, Whaley J, Swindlehurst C, McCormack R, Wolfert R, Selkoe D J, Lieberburg I, Schenk D. Nature (London) 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 9.Shoji M, Golde T E, Ghiso J, Cheung T T, Estus S, Shaffer L M, Cai X, McKay D M, Tintner R, Frangione B, Younkin S G. Science. 1992;258:126–129. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- 10.Jarrett J T, Berger E P, Lansbury P T., Jr Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 11.Dovey H F, Suomensaari-Chrysler S, Lieberburg I, Sinha S, Keim P S. Neuroreport. 1993;4:1039–1042. doi: 10.1097/00001756-199308000-00011. [DOI] [PubMed] [Google Scholar]
- 12.Asami-Odaka A, Ishibashi Y, Kikuchi T, Kitada C, Suzuki N. Biochemistry. 1995;34:10272–10278. doi: 10.1021/bi00032a022. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki N, Cheung T T, Cai X-D, Odaka A, Otvos L, Jr, Eckman C, Golde T E, Younkin S G. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 14.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird T D, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S G. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 15.Higaki J, Quon D, Zhong Z, Cordell B. Neuron. 1995;14:651–659. doi: 10.1016/0896-6273(95)90322-4. [DOI] [PubMed] [Google Scholar]
- 16.Selkoe D J, Podlisny M B, Joachim C L, Vickers E A, Lee G, Fritz L C, Oltersdorf T. Proc Natl Acad Sci USA. 1988;85:7341–7345. doi: 10.1073/pnas.85.19.7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung A Y, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe D J. Nature (London) 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 18.Haass C, Schlossmacher M G, Hung A Y, Vigo-Pelfrey C, Mellon A, Ostaszewski B L, Lieberburg I, Koo E H, Schenk D, Teplow D B, Selkoe D J. Nature (London) 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 19.Haass C, Hung A Y, Selkoe D J, Teplow D B. J Biol Chem. 1994;269:17741–17748. [PubMed] [Google Scholar]
- 20.Köhler G, Milstein C. Eur J Immunol. 1976;6:511–519. doi: 10.1002/eji.1830060713. [DOI] [PubMed] [Google Scholar]
- 21.Saido T C, Iwatsubo T, Mann D M A, Shimada H, Ihara Y, Kawashima S. Neuron. 1995;14:457–466. doi: 10.1016/0896-6273(95)90301-1. [DOI] [PubMed] [Google Scholar]
- 22.Podlisny M B, Tolan D, Selkoe D J. Am J Pathol. 1991;138:1423–1435. [PMC free article] [PubMed] [Google Scholar]
- 23.Knops J, Suomensaari S, Lee M, McConlogue L, Seubert P, Sinha S. J Biol Chem. 1995;270:2419–2422. doi: 10.1074/jbc.270.6.2419. [DOI] [PubMed] [Google Scholar]
- 24.Haass C, Lemere C A, Capell A, Citron M, Seubert P, Schenk D, Lannfelt L, Selkoe D J. Nat Med. 1995;1:1291–1296. doi: 10.1038/nm1295-1291. [DOI] [PubMed] [Google Scholar]
- 25.Citron M, Teplow D B, Selkoe D J. Neuron. 1995;14:661–670. doi: 10.1016/0896-6273(95)90323-2. [DOI] [PubMed] [Google Scholar]
- 26.Citron M, Diehl T S, Capell A, Haass C, Teplow D B, Selkoe D J. Neuron. 1996;17:171–179. doi: 10.1016/s0896-6273(00)80290-1. [DOI] [PubMed] [Google Scholar]
- 27.Hung A Y, Haass C, Nitsch R M, Qiu W Q, Citron M, Wurtman R J, Growdon J H, Selkoe D J. J Biol Chem. 1993;268:22959–22962. [PubMed] [Google Scholar]
- 28.Buxbaum J D, Koo E H, Greengard P. Proc Natl Acad Sci USA. 1993;90:9195–9198. doi: 10.1073/pnas.90.19.9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gravina S A, Ho L, Eckman C B, Long K E, Otvos L J, Younkin L H, Suzuki N, Younkin S G. J Biol Chem. 1995;270:7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 30.Koo E H, Squazzo S. J Biol Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- 31.Selkoe D J. Annu Rev Cell Biol. 1994;10:373–403. doi: 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- 32.Haass C, Hung A Y, Schlossmacher M G, Teplow D B, Selkoe D J. J Biol Chem. 1993;268:3021–3024. [PubMed] [Google Scholar]
- 33.Haass C, Capell A, Citron M, Teplow D B, Selkoe D J. J Biol Chem. 1995;270:6186–6192. doi: 10.1074/jbc.270.11.6186. [DOI] [PubMed] [Google Scholar]
- 34.Sandhu F A, Kim Y, Lapan K A, Salim M, Aliuddin V, Zain S B. Proc Natl Acad Sci USA. 1996;93:2180–2185. doi: 10.1073/pnas.93.5.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maruyama K, Terakado K, Usami M, Yoshikawa K. Nature (London) 1990;347:566–569. doi: 10.1038/347566a0. [DOI] [PubMed] [Google Scholar]
- 36.Yankner B A, Dawes L R, Fisher S, Villa-Komaroff L, Oster-Granite M L, Neve R L. Science. 1989;245:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
- 37.Lemere C A, Blusztajn J K, Yamaguchi H, Wisniewski T, Saido T C, Selkoe D J. Neurobiol Dis. 1996;3:16–32. doi: 10.1006/nbdi.1996.0003. [DOI] [PubMed] [Google Scholar]