

Abstract
Nitric oxide produced in endothelial cells affects vascular tone. To investigate the role of endothelial nitric oxide synthase (eNOS) in blood pressure regulation, we have generated mice heterozygous (+/−) or homozygous (−/−) for disruption of the eNOS gene. Immunohistochemical staining with anti-eNOS antibodies showed reduced amounts of eNOS protein in +/− mice and absence of eNOS protein in −/− mutant mice. Male or female mice of all three eNOS genotypes were indistinguishable in general appearance and histology, except that −/− mice had lower body weights than +/+ or +/− mice. Blood pressures tended to be increased (by approximately 4 mmHg) in +/− mice compared with +/+, while −/− mice had a significant increase in pressure compared with +/+ mice (≈18 mmHg) or +/− mice (≈14 mmHg). Plasma renin concentration in the −/− mice was nearly twice that of +/+ mice, although kidney renin mRNA was modestly decreased in the −/− mice. Heart rates in the −/− mice were significantly lower than in +/− or +/+ mice. Appropriate genetic controls show that these phenotypes in F2 mice are due to the eNOS mutation and are not due to sequences that might differ between the two parental strains (129 and C57BL/6J) and are linked either to the eNOS locus or to an unlinked chromosomal region containing the renin locus. Thus eNOS is essential for maintenance of normal blood pressures and heart rates. Comparisons between the current eNOS mutant mice and previously generated inducible nitric oxide synthase mutants showed that homozygous mutants for the latter differ in having unaltered blood pressures and heart rates; both are susceptible to lipopolysaccharide-induced death.
Keywords: gene disruption, heart rate, renin concentration, lipopolysaccharide
Nitric oxide (NO) is produced from arginine and oxygen in a variety of mammalian cell types by three distinct nitric oxide synthase (NOS) isozymes: two constitutively transcribed forms, neuronal (designated nNOS below) (1, 2) and endothelial (designated eNOS below) (3–5) enzymes, and an inducible form (designated iNOS below) found in a number of cell types, including macrophages and vascular smooth muscle cells (6–9). NO produced in endothelial cells is a vasodilator and antithrombogenic agent (10, 11). In response to agents such as bradykinin and acetylcholine or to mechanical forces such as shear stress, endothelial cells produce NO, which diffuses into vascular smooth muscle cells and causes vasorelaxation. Arginine analogs, such as NG-monomethyl-l-arginine and l-nitroarginine, are competitive inhibitors of NOS enzymes. Administration of these compounds to animals (10, 12–15) or to humans (16, 17) results in elevated blood pressure and increased peripheral resistance. Response to NG-monomethyl-l-arginine has been reported to be abnormal in hypertensive patients compared with normotensive individuals, with a lowered response being correlated with higher blood pressure (18). Thus the NOS isozymes and their genes are candidates for involvement in hypertension, although it has not been possible to prove the specific involvement of eNOS because the currently available NOS inhibitors affect all three isozymes. To specifically study the role of NO produced by eNOS in blood pressure regulation, we have generated mice that lack a functional endothelial nitric oxide gene (eNOS). To further characterize the specific influence of endothelium-derived NO, we also compared the eNOS mutant mice to our previously reported iNOS mutant mice (19).
MATERIALS AND METHODS
Cloning of a Murine eNOS Genomic Fragment.
An approximately 450-bp fragment extending from within exon 12 of the eNOS gene to within exon 13 was amplified from bovine genomic DNA by PCR. The primers had the following sequences based on data from bovine eNOS cDNA sequences, 5′-ATGGCCAAGCGAGTGAAAGCAACCAT-3′ and 5′-GCATTCCCAAAGGTGCTGGTCACCAC-3′ (20). The resulting PCR fragment hybridized to a 4.7-kb band in Southern blots of XbaI-digested murine strain 129/Ola (129) genomic DNA. When the PCR fragment was used as a probe to screen a λ bacteriophage Charon 35 library having inserts from an XbaI digestion of murine strain 129 genomic DNA, a clone containing a 4.7-kb XbaI fragment was obtained. DNA sequence analysis of a portion of this fragment showed an 89% nucleotide sequence identity to human eNOS exon sequences with an identical placement of intron 12 (21), compared with 60% sequence identity to rat neuronal NOS cDNA (the closest isoform to eNOS) (2) (data not shown).
Targeting Construct.
An eNOS targeting construct, pENOSX (Fig. 1A), was prepared from the 4.7-kb XbaI fragment by the replacement of 129 bp in exon 12 with 1.2 kb of sequences that include the neomycin-resistance gene (neo) from pMC1Neo polA (22) oriented oppositely to the eNOS gene. This replacement disrupts the calmodulin binding site essential to eNOS function and introduces a premature translation stop codon into the eNOS transcripts. The vector plasmid was the herpes simplex virus thymidine kinase-containing plasmid pPNT (23) modified by deletion of its neo gene.
Figure 1.
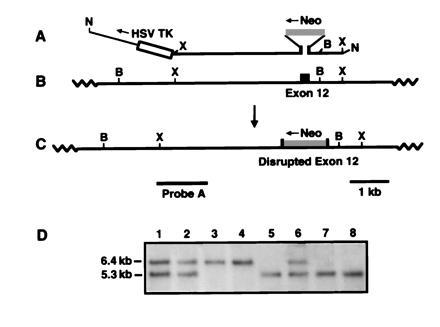
eNOS targeting strategy. (A) Targeting construct pENOSX. The heavy line represents eNOS genomic sequences with the disrupted exon 12 indicated by solid bars. Shaded and open boxes represent the neo (neomycin resistance) gene and herpes simplex virus thymidine kinase genes, respectively. The thin line is plasmid vector (not to scale). (B) Portion of the endogenous murine eNOS gene. (C) Correctly targeted eNOS gene. (D) Southern blot, hybridized to probe A (indicated by a horizontal bar), of BamHI digested genomic DNA from offspring of an eNOS +/− × eNOS +/− mating. The 5.3-kb band indicates a wild-type eNOS gene while the 6.4-kb band indicates a disrupted eNOS gene. B, BamHI; N, NotI; X, XbaI.
Embryonic Stem (ES) Cell Growth and Transformation.
Murine ES cells from subclone BK4 of line E14TG2a (24) were cultured as described (25). Electroporation of ≈3 × 107 cells in 0.5 ml of growth medium was carried out with NotI-linearized pENOSX at a concentration of 2 nM using a 1-sec discharge from a 250-μF capacitor charged to 300 V. The cells were then cultured on murine embryonic fibroblast feeder cells in 10-cm-diameter tissue culture dishes. The following day, the medium was replaced with medium containing G418 at 200 μg/ml and ganciclovir at 2 μg/ml. Clones resistant to both drugs were individually dispersed into 1.5-cm-diameter tissue culture wells with feeder cells. On reaching confluence they were dispersed to larger plates with feeder cells for further growth, as well as to 3.5-cm wells without feeder cells for use in preparing genomic DNA. Southern blot analysis of ES cell genomic DNA with various restriction enzymes and probes was used to confirm the targeting event.
Generation of eNOS-Deficient Mice.
Chimeras were generated from ES cells carrying the disrupted eNOS gene by injection into C57BL/6J (B6) blastocysts followed by transfer to pseudopregnant CD1 females for completion of development. Male chimeras mated to B6 females generated B6/129 F1 hybrids that were either wild-type (+/+) or heterozygous (+/−) for the disrupted eNOS gene. F1 +/− mice were intercrossed to generate F2 offspring of all three genotypes, +/+, +/−, and −/−; these F2 mice were used for the experiments described herein. Determination of mouse genotypes was by Southern blot analysis of genomic tail DNA using probe A (see Fig. 1); probe A is a genomic DNA RsaI fragment from the 5′ end of the cloned XbaI eNOS gene fragment.
Immunohistochemical Localization of eNOS.
Hearts and kidneys were rapidly removed and embedded in OCT compound (Miles) for preparing 7-μm frozen sections. Sections were fixed with acetone (−20°C at start) for 10 min at 4°C, washed twice with phosphate-buffered saline containing 0.1 g of bovine serum albumin per 100 ml (PBS/0.1% BSA) at room temperature. Sections were then incubated with 0.3% hydrogen peroxide in methanol for 30 min at room temp to block endogenous peroxidase activity and washed again with PBS/0.1% BSA. Nonspecific antibody binding was blocked by incubation for 4–5 h with PBS/4% BSA. Sections were then washed twice with PBS/0.1% BSA and incubated 14–16 h with a 1:1000 dilution of an eNOS-specific polyclonal antiserum (Transduction Laboratories, Lexington, KY). Sections were washed and developed with the Vectastain ABC system (Vector Laboratories) using diaminobenzidine tetrahydrochloride. Sections were counterstained with hematoxylin, mounted in Permamount, and examined by light microscopy.
Histological Analysis.
Tissue samples were collected immediately after euthanization and were fixed in 10% neutral-buffered formalin. Sections were stained with hematoxylin/eosin.
Blood Pressure and Heart Rate Determination.
Tail-cuff blood pressures and heart rates were determined in mice aged between 90 and 110 days in “blinded” fashion using a computerized tail-cuff system (Visitech Systems, Cary, NC) as described (26).
Plasma Renin Concentration and Kidney Renin mRNA Concentration.
Arterial blood samples were rapidly withdrawn from eNOS −/− and age matched +/+ sibs under CO2 anesthesia (less than 1 min from loss of consciousness to the end of collection) using noncoated glass pipettes into ice-cold microcentrifuge tubes containing EDTA and immediately centrifuged to isolate plasma. Plasma renin concentrations were determined by a radioimmunoassay for angiotensin I as described (27).
Kidney tissues were dissected from mice after withdrawing the blood samples. Total RNA was isolated as described (28). RNase protection assays were as described by Azrolan and Breslow (29) with the RNA probe being a 290-bp murine renin exon 9 fragment (30).
Lipopolysaccharide (LPS) Treatment.
Mice were injected i.p. with bacterial LPS (Escherichia coli, serotype 026:B6, Difco) at 12.5 mg/kg and monitored over a 96-h period.
Statistical Analysis.
Data are presented as mean ± SEM. Genotype frequencies were tested for deviation from Mendelian expectations by χ2 tests. For comparisons, we used one or two factor analysis of variance with pairwise comparisons by Tukey’s test.
RESULTS
Disruption of the Murine eNOS Gene by Gene Targeting.
The gene targeting strategy used to disrupt the eNOS gene (insertion of the neo gene accompanied by a partial gene deletion) results in disruption of the calmodulin-binding site and the introduction of a premature translational stop site (Fig. 1). This deletion/disruption of the calmodulin-binding site was expected to inactivate eNOS, since calmodulin binding is required for electron transfer to the active site of the enzyme. The linearized targeting construct (pENOSX) was electroporated into murine ES cells and clones resistant to both G418 and ganciclovir were obtained. Targeted cells were identified by Southern blot analysis of genomic DNA from the doubly resistant clones using various enzyme digests and probes (data not shown). In a total of 87 doubly resistant clones screened, 18 were correctly targeted.
Generation of eNOS-Deficient Mice.
Targeted ES cells were injected into B6 blastocysts which were transferred into pseudopregnant CD1 mice for implantation and development. Resulting male chimeras were bred to B6 females to generate F1 129/B6 offspring.
F1 mice heterozygous for the disrupted eNOS gene were intercrossed to generate F2 offspring that were genotyped by Southern blot analysis of tail genomic DNA digested with BamHI and hybridized to probe A (Fig. 1). The wild-type allele gives a 5.3-kb band under these conditions, while the targeted allele yields a 6.4-kb band (Fig. 1D). Analysis of 288 F2 mice revealed that 67 (23%) were wild-type (+/+), 169 (59%) were heterozygous (+/−), and 52 (18%) were homozygous mutant (−/−). The overall distribution deviates somewhat from a 1:2:1 Mendelian distribution (P < 0.01) with the major deviation being due to a deficiency in homozygous mutants.
Absence of eNOS Protein in −/− Mice.
Immunohistochemical tests for eNOS protein, using polyclonal eNOS-specific antibodies were performed on frozen sections of heart and kidney prepared from mice of each eNOS genotype. Sections from +/+ mice show staining in most cardiac capillary endothelial cells indicated by arrowheads and in renal glomerular tufts, while sections from −/− show essentially no staining (Fig. 2). This confirms that the −/− mice lack detectable eNOS. Mice +/− for the eNOS mutation showed reduced but positive staining when compared with +/+ mice (data not shown). Controls with secondary antibody and diaminobenzidine were completely negative. Identical positive results were obtained with another eNOS polyclonal antiserum raised against a different epitope (data not shown).
Figure 2.
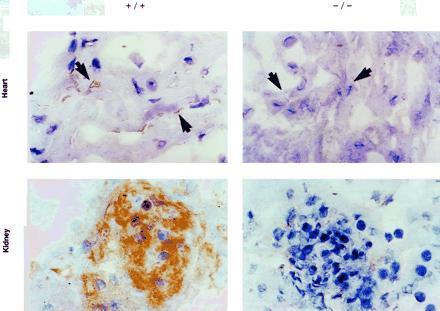
Immunohistochemical localization of eNOS. Heart and kidney sections from eNOS +/+ and −/− animals incubated with an anti-eNOS polyclonal antiserum, washed, and developed with the Vectastain ABC system using diaminobenzidine. The +/+ mice show intense staining in capillaries surrounding individual myocytes (arrowheads) (Upper Left) and in glomerular tufts (Lower Left). Staining is virtually absent in the capillaries (arrowheads) (Upper Right) or glomerular tufts (Lower Right) of the −/− mice. No eNOS staining is visible in the cardiac myocytes of either genotype.
General Appearance, Fertility, Body Weight, and Histology.
The −/− and +/− mice are indistinguishable from normal (+/+) in general appearance, and both are fertile. However, a comparison of body weights at approximately 14 weeks showed that the effect of eNOS genotype (P < 0.01) and gender (P < 0.001) were statistically significant although the interaction of genotype with gender was not (P = 0.18). Inspection of the data revealed that three mice were outliers, having weights differing by more than 2 SD from the means of their groups. Exclusion of these three mice and reanalysis of the data gave the same significance for genotype and gender with −/− mice having an approximately 7.5% lower body weight than +/+. Weights for females were as follows: −/−, 21.9 ± 0.6 g (n = 10); +/−, 23.9 ± 0.4 g (n = 28); +/+, 24.1 ± 0.5 g (n = 17). Weights for males were as follows: −/−, 27.3 ± 0.6 g (n = 7); +/−, 30.6 ± 0.7 g (n = 19); +/+, 29.0 ± 0.7 g (n = 12).
Histological examination of heart, liver, lung, aorta, kidney, brain, spleen, and adrenal glands from −/− and +/− F2 mice showed no obvious differences from wild-type F2 mice. The heart weight to body weight ratios of the −/− and +/+ mice were not significantly different.
Analysis of Blood Pressure in Wild-Type and Mutant Mice.
To evaluate the role of eNOS in blood pressure regulation, we determined the tail-cuff blood pressure for F2 mice of all three eNOS genotypes and both sexes (Fig. 3A). The effect of eNOS genotype was highly significant (P < 10−8) while the effect of gender (P = 0.84) and the interaction of gender and genotype (P = 0.73) were not. Blood pressures of +/− mice (125.6 ± 1.5 mmHg, n = 38) tended to be higher than those of +/+ mice (121.8 ± 1.8 mmHg, n = 34), but the difference did not reach statistical significance (P = 0.20). The blood pressures of −/− mice (140.3 ± 2.3 mmHg, n = 21) were significantly higher than the pressures of either the +/− mice (P = 1 × 10−4) or the +/+ mice (P = 1 × 10−4).
Figure 3.
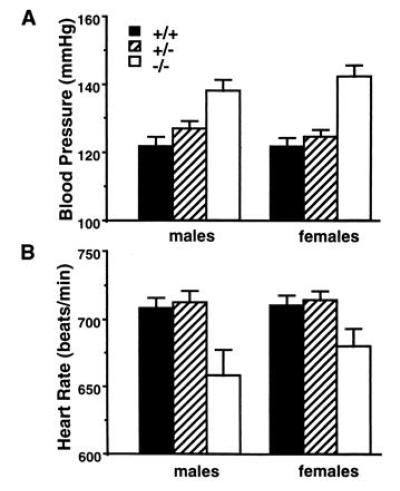
Effect of eNOS genotypes (+/+, +/−, and −/−) on blood pressure (A) and heart rate (B). Error bars indicate SEM. Numbers of mice are as follows: 34, +/+; 38, +/−; 21, −/−.
Analysis of Heart Rate in Wild-Type and Mutant Mice.
We also evaluated the heart rates of F2 mice of all three eNOS genotypes and both sexes (Fig. 3B). The effect of eNOS genotype was significant (P < 1 × 10−4) while the effect of gender (P = 0.30) and the interaction of genotype and gender (P = 0.57) were not. The −/− mice have a significantly reduced heart rate (670 ± 11 beats per min) compared with either the +/+ (709 ± 5 beats per min; P < 0.001) or +/− mice (713 ± 5; P < 0.001).
Controls for Linked and Unlinked Genes.
In experiments involving F2 mice, it is important to note that observed phenotypic differences may arise (i) from the genotype at the target locus, (ii) from inheritance of sequences linked to the target gene that differ between B6 and 129 and affect the phenotype under investigation, and (iii) from segregation of unlinked sequences that is by chance biased in some direction which affects the phenotype.
All our F2 mice derive their disrupted eNOS genes together with linked sequences from strain 129 and their wild-type eNOS genes and linked sequences from strain B6. Thus the possibility exists that gene(s) linked to eNOS and differing in strains 129 and B6 could contribute to the observed phenotypic differences among the −/−, +/−, and +/+ mice. To test for effects of sequences linked to the eNOS gene, we compared the phenotypes of 109 control F2 mice inheriting two B6 (B6/B6), one B6 and one 129 (B6/129), or two 129 (129/129) wild-type eNOS alleles. The control F2 mice used for this purpose were either wild-type or had mutations in the unlinked gene coding for angiotensin-converting enzyme that do not significantly affect blood pressure. The control mice were genotyped for the source of their eNOS alleles by Southern blots hybridized to probe A using DNA digested with DraI; this generates an 8-kb band from the B6-derived eNOS gene and a 14-kb band from 129-derived eNOS gene.
Tail-cuff blood pressure measurements showed no blood pressure differences between the control F2 mice having their wild-type eNOS genes derived from B6/B6 (118.1 ± 1.9 mmHg, n = 31), B6/129 (117.1 ± 1.6 mmHg, n = 51), or 129/129 (118.0 ± 1.9 mmHg, n = 27); P = 0.96 for B6/B6 versus 129/129. Similar absences of effects of any linked genes were obtained in comparisons of heart rates and body weights in these controls (data not shown). Since in each case there are no phenotypic differences between the control F2 mice that have B6 and/or 129 wild-type eNOS genes, we conclude that the observed phenotypic differences between the experimental eNOS −/− and +/+ mice are due to the disrupted eNOS gene itself and not to any differences between linked genes in the two strains.
Segregation of important unlinked sequences that by chance is biased may also influence the results of experiments involving F2 mice, although in general the probability of this occurring is reduced by studying increasing numbers of animals. In our experiments, for example, the renin genes of strains B6 and 129 differ (Ren-1c for B6; Ren-1d and Ren-2 for 129). We genotyped 81 of our F2 mice for this known polymorphism between the two strains. The results showed that within each eNOS genotype (+/+, +/−, −/−) all three renin genotypes were represented at frequencies not differing significantly from Mendelian expectations. This demonstrates that the number of mice examined was sufficient for achieving an unbiased segregation of this particular unlinked gene.
Analysis of Renin in Wild-Type and Mutant Mice.
Total RNA was prepared from the kidneys of F2 +/+ and −/− mice and the amount of renin mRNA was determined by an RNase protection assay (Fig. 4A). The effect of genotype (P = 0.07) trended toward significance, while gender was significant (P = 0.003), but there was no interaction of genotype and gender (P = 0.87). Renin mRNA was slightly reduced in the −/− mice (females 18.5 ± 1.4 pg/μg, n = 4; males 14.2 ± 0.8 pg/μg, n = 4) compared with +/+ mice (females 20.6 ± 1.5 pg/μg, n = 5; males 16.7 ± 0.5 pg/μg, n = 5). Expressed as a percentage of the mean of like-sexed +/+ (100 ± 3.8%) mice, the renin mRNA levels in the combined male and female −/− animals was 87.5 ± 3.9% (P = 0.04).
Figure 4.
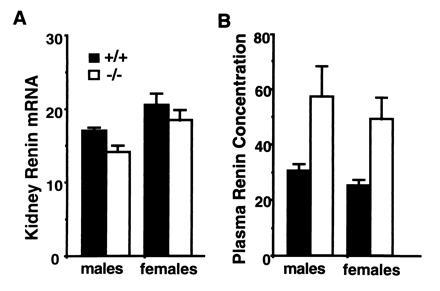
Effect of eNOS genotypes (+/+ and −/−) on kidney renin mRNA (A) and plasma renin concentration (B). Kidney renin mRNA units are pg of mRNA per μg of total RNA. Plasma renin concentration units are angiotensin I production (ng/hr) per ml of plasma. Error bars indicate SEM. Numbers of mice are as follows: 5 male and 5 female, +/+; 4 male and 4 female, −/−.
In contrast, as shown in Fig. 4B, plasma renin concentrations in the same mice, determined as the rate of angiotensin I production in the presence of excess angiotensinogen, were nearly twice as high in the −/− mice (females 49.9 ± 7.7 ng per ml per hr and males 57.8 ± 11.1 ng per ml per hr) as in the +/+ mice (females 25.7 ± 2.0 ng per ml per hr and males 30.8 ± 2.7 ng per ml per hr). The effect of eNOS genotype was statistically significant (P = 0.001) while the effect of gender (P = 0.31) and the interaction of genotype and gender (P = 0.82) were not significant. Examination of the renin genotypes of the 18 F2 animals used for the renin measurements showed that differences in the renin concentrations in the individual eNOS +/+ and −/− mice could not be explained by their renin genotypes.
Comparisons Between iNOS and eNOS Mutant Mice.
We have reported (19) the phenotype of mice lacking a functional iNOS gene.
To determine whether this isoform of NOS also affects blood pressure, we measured the tail-cuff blood pressures of the iNOS mutants. Overall, the effect of iNOS genotype was not significant (P = 0.8). There were no significant differences between −/− (113.8 ± 2.5 mmHg, n = 12), +/− (115.0 ± 2.6 mmHg, n = 12), or +/+ (116.3 ± 2.9 mmHg, n = 11), with P > 0.75 for all pairwise comparisons. We also established that any strain differences in loci linked to iNOS did not affect blood pressure (P = 0.12) using the same 109 F2 control mice described above. Thus absence of iNOS (unlike absence of eNOS) leads to no differences in blood pressure. Similarly, no statistically significant differences in the heart rates of the iNOS mutants were observed.
A further comparison was made between the eNOS and iNOS mutants because our data (19) had shown unexpectedly that iNOS −/− mice die from injected LPS at the same frequency as iNOS +/+ mice. This raised the possibility that eNOS rather than iNOS is important in septic shock. To test this possibility, we injected mice of each eNOS genotype with LPS at 12.5 mg/kg i.p. (a dose shown to produce about 75% deaths in our previous work) and monitored them over a 96-h period. The results with the different eNOS genotypes are essentially identical as shown in Fig. 5. Eighty percent of +/+ (8 of 10), 70% of +/− (7 of 10), and 80% of −/− (8 of 10) mice died by 96 h. Thus absence of eNOS (like absence of iNOS) activity does not protect mice from death following LPS administration.
Figure 5.
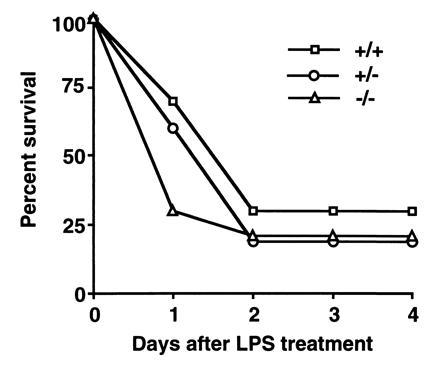
Survival of LPS-treated mice. Ten F2 mice of each eNOS genotype (+/+, +/−, and −/−) were treated i.p. with LPS at 12.5 mg/kg and survival was monitored over 4 days.
DISCUSSION
We have inactivated the murine eNOS gene by homologous recombination in ES cells and have generated mice of the three eNOS genotypes (+/+, +/−, and −/−). The homozygous mutants completely lack eNOS and are grossly and histologically normal, although they have significantly lower body weights. We do not at present have any additional information relevant to this difference in body weights.
Our main aim in generating the eNOS −/− mice was to investigate the effects of the endothelial enzyme on blood pressure. We expected that mice lacking endothelium-derived NO would have elevated blood pressures. This expectation was confirmed by tail-cuff blood pressure measurements that demonstrate that conscious F2 −/− mice of either sex have significantly elevated pressures (about 18 mmHg above their wild-type sibs and 14 mmHg above their +/− sibs). These results are in agreement with the recent report of blood pressure measurements made using intraarterial catheterization on eNOS −/− and +/+ F2 129/B6 mice generated independently by Huang et al. (31). Our heterozygotes for the eNOS disruption showed a tendency toward increase in blood pressure over wild type, but the difference did not reach statistical significance. This suggests that sufficient eNOS is produced from a single copy of the eNOS gene to maintain near normal pressures or that other systems involved in blood pressure regulation compensate almost completely for the partial lack of eNOS. Whether or not the heterozygotes would show a significant difference from wild type in other environmental or genetic backgrounds remains to be determined.
In addition to the increases in blood pressure, we observed a statistically significant reduction in the heart rates of the eNOS −/− mice compared with either +/+ or +/− mice. This is in contrast to the report by Huang et al. (31) that the heart rates of their eNOS mutant mice and wild-type mice did not differ. The difference between these two observations may be that our heart rate measurements were made on conscious mice, while Huang et al. (31) used urethane-anesthetized mice. Reduced heart rates in conscious rats (32), dogs (33), and humans (34) administered a NOS inhibitor have been reported, with the authors concluding that the most likely cause was baroreceptor reflex mechanisms responding to elevated blood pressure. Interestingly, while our −/− mice have elevated blood pressures and reduced heart rates, their hearts appear normal histologically and their heart/body weight ratios do not differ from those of +/+ mice.
An important element in our present study is the effective elimination of potential complications introduced by the use of F2 hybrids between the two different mouse strains (129 and B6) in the eNOS experiments. Because the phenotypes studied are all quantitative in nature, it is essential to establish that they are due to the disrupted eNOS genes and not to inadvertent effects of other linked or unlinked gene differences between the strains. To evaluate whether the phenotypes in our F2 mice are the result of genes linked to the eNOS locus, we determined the phenotypes of control F2 mice having wild-type eNOS genes derived from 129 and/or B6 and found that body weights, blood pressures, and heart rates were not affected by the source of the wild-type eNOS genes and of genes linked to the eNOS locus. Thus, we conclude that the phenotypic alterations seen in blood pressure, heart rate, and body weight in −/− mice are caused by the disruption of the eNOS gene itself.
The possibility of a chance bias in the segregation of genes unlinked to the eNOS locus influencing results can be reduced in the general case by ensuring the use of sufficient numbers of F2 animals to achieve random segregation of any relevant genes. In specific cases, tests can be made to determine whether strain differences in any unlinked genes of particular concern have been inherited in an unbiased fashion. Our observations on the segregation of the B6/129 strain differences at the renin locus illustrate both points: all three renin genotypes were represented among the mice of the three eNOS genotypes in proportions that did not differ from Mendelian expectations, and renin genotypes could not explain the differences in renin concentrations of 18 mice tested.
There are at least two potential mechanisms by which absence of eNOS may result in an increase in blood pressure. The most obvious is that the eNOS −/− mice lack the vasorelaxing pathway mediated by endothelium-derived NO on vascular smooth muscle. Our data (Fig. 2) on the absence of eNOS in endothelial walls of the −/− mice supports this possibility. A second possible mechanism, not excluded by the first, is that NO plays a role in the release of renin from the kidney into the bloodstream. Past work on the role of NO in regulating renin release has yielded conflicting conclusions. In vivo and in vitro experiments have led some authors to conclude that NO suppresses renin release (35–37), while others have concluded that NO stimulates renin release (38–40). None of these experiments was able to fully isolate the function of the individual NOS isotypes. However, our eNOS −/− mice provide the opportunity to address this possibility in mice that lack only the eNOS isoform. As shown in Fig. 4B, we found that the −/− mice have nearly twice the plasma renin concentration of the +/+ mice. This result suggests that at least part of the increased blood pressures in the eNOS −/− mice could be due to their having higher than normal plasma renin concentrations that would be expected to lead to an increased rate of angiotensin II production.
The higher than normal plasma renin concentration in the eNOS −/− mice is, however, paradoxical for two reasons. (i) An increased blood pressure, as seen in these mice, would be expected to lead to a homeostatic reduction in renin secretion due to the renal baroreceptor mechanism rather than to an increase. (ii) The high plasma renin concentration is accompanied by a modest decrease in kidney renin mRNA (Fig. 4A). At present we cannot exclude the possibility that an acute release of stored renin is induced during the blood drawing, but this response would have to be markedly enhanced in the −/− mice. Clearly further investigation of the role of eNOS in renin production and release is warranted.
Several explanations are possible for the reduction in heart rate that is seen in the eNOS −/− mice. One possibility is that the baroreceptor reflex is (indirectly) reset as a consequence of the chronic increase in blood pressure in these mice. Against this explanation is the observation that mice lacking atrial natriuretic peptide have approximately equal increases in their blood pressures but do not have heart rates statistically different from control mice (41). A second possibility is that NO produced by eNOS could play a direct role in the modulation of heart rates. However, at the present we are unable to specify the exact cause of the reduced heart rate in eNOS −/− mice.
NO has been thought to play an important role in the occurrence of death during septic shock (42–44). Because we have observed (19) that mice lacking the iNOS isoform are not protected from LPS-induced death, we tested whether lack of the eNOS isoform might be protective. We found that mice lacking eNOS died at the same frequency as wild-type animals, and we conclude that neither the production of NO by eNOS nor by iNOS is essential for death in this model of septic shock. Additionally, while our current data clearly demonstrate effects of eNOS mutations on blood pressure, we did not find any effect of iNOS mutations on blood pressure.
In summary, our study shows the eNOS −/− mice have three differences from wild type: an increased blood pressure, an elevation of plasma renin concentration despite a modest decrease in kidney renin mRNA, and a reduced heart rate. We conclude that eNOS but not iNOS is essential for the maintenance of normal blood pressure.
Acknowledgments
We thank Sylvia Hiller, Kimberly Kluckman, Rob Murray, Annette Staton, and Gloria O’Neal for their technical help and Drs. William Beirwaltes and Tom Coffman for valuable discussions. This work was supported by National Institutes of Health Grants GM20069 (O.S.), HL49277 (O.S.), HL03470 (J.H.K.), and HL51948 (W.C.S.) and by a Wellcome Trust Grant 8038282/8/93 (K.M.D.). J.H.K. was a Howard Hughes Physician Research Fellow, and W.C.S. is an Established Investigator of the American Heart Association. The University of North Carolina gratefully acknowledges support from the W. M. Keck Foundation.
Footnotes
Abbreviations: ES cell, embryonic stem cell; neo, neomycin phosphotransferase; LPS, lipopolysaccharide; eNOS and iNOS, endothelial and inducible nitric oxide synthase, respectively.
References
- 1.Bredt D S, Hwang P M, Snyder S H. Nature (London) 1990;347:768–770. doi: 10.1038/347768a0. [DOI] [PubMed] [Google Scholar]
- 2.Bredt D S, Hwang P M, Glatt C E, Lowenstein C L, Reed R R, Snyder S H. Nature (London) 1991;351:714–718. doi: 10.1038/351714a0. [DOI] [PubMed] [Google Scholar]
- 3.Pollock J S, Förstermann U, Mitchell J A, Warner T D, Schmidt H H H W, Nakane M, Murad F. Proc Natl Acad Sci USA. 1991;88:10480–10484. doi: 10.1073/pnas.88.23.10480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janssens S P, Shimouchi A, Quertermous T, Bloch D B, Bloch K D. J Biol Chem. 1992;267:14519–14522. [PubMed] [Google Scholar]
- 5.Sessa W C, Harrison J K, Barber C M, Zeng D, Durieux M E, D’Angelo D D, Lynch K R, Peach M J. J Biol Chem. 1992;267:15274–15276. [PubMed] [Google Scholar]
- 6.Stuehr D J, Cho H J, Kwon N S, Weise M, Nathan C F. Proc Natl Acad Sci USA. 1991;88:7773–7777. doi: 10.1073/pnas.88.17.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nussler A K, Di Silvio M, Billiar T R, Hoffman R A, Geller D A, Selby R, Madariaga J, Simmons R L. J Exp Med. 1992;176:261–264. doi: 10.1084/jem.176.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Charles I G, Palmer R M J, Hickery M S, Bayliss M T, Chubb A P, Hall V S, Moss D W, Moncada S. Proc Natl Acad Sci USA. 1993;90:11419–11423. doi: 10.1073/pnas.90.23.11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott-Burden T, Schini V B, Elizondo E, Junquero D C, Vanhoutte P M. Circ Res. 1992;71:1088–1100. doi: 10.1161/01.res.71.5.1088. [DOI] [PubMed] [Google Scholar]
- 10.Rees D D, Palmer R M J, Moncada S. Proc Natl Acad Sci USA. 1989;86:3375–3378. doi: 10.1073/pnas.86.9.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aisaka K, Gross S S, Griffith O W, Levi R. Biochem Biophys Res Commun. 1989;160:881–886. doi: 10.1016/0006-291x(89)92517-5. [DOI] [PubMed] [Google Scholar]
- 12.Whittle B J, Lopez-Belmonte J, Rees D D. Br J Pharmacol. 1989;98:646–652. doi: 10.1111/j.1476-5381.1989.tb12639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salazar F J, Pinilla J M, Lopez F, Romero J C, Quesada T. Hypertension. 1992;20:113–117. doi: 10.1161/01.hyp.20.1.113. [DOI] [PubMed] [Google Scholar]
- 14.Baylis C, Mitruka B, Deng A. J Clin Invest. 1992;90:278–281. doi: 10.1172/JCI115849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ribeiro M O, Anyunes E, de Nucci G, Lovisolo S M, Zatz R. Hypertension. 1992;20:298–303. doi: 10.1161/01.hyp.20.3.298. [DOI] [PubMed] [Google Scholar]
- 16.Vallance P, Collier J, Moncada S. Cardiovasc Res. 1989;23:1053–1057. doi: 10.1093/cvr/23.12.1053. [DOI] [PubMed] [Google Scholar]
- 17.Vallance P, Collier J, Moncada S. Lancet. 1989;ii:997–1000. doi: 10.1016/s0140-6736(89)91013-1. [DOI] [PubMed] [Google Scholar]
- 18.Calver A, Collier J, Moncada S, Vallance P. J Hypertens. 1992;10:1025–1031. [PubMed] [Google Scholar]
- 19.Laubach V E, Shesely E G, Smithies O, Sherman P A. Proc Natl Acad Sci USA. 1995;92:10688–10692. doi: 10.1073/pnas.92.23.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamas S, Marsden P A, Tempest P, Michel T. Proc Natl Acad Sci USA. 1992;89:6348–6352. doi: 10.1073/pnas.89.14.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marsden P A, Heng H H, Scherer S W, Stewart R J, Hall A V, Shi X-M, Tsui L-C, Schappert K T. J Biol Chem. 1993;268:17478–17488. [PubMed] [Google Scholar]
- 22.Thomas K R, Capecchi M R. Cell. 1987;51:503–512. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- 23.Tybulewicz V L J, Crawford C E, Jackson P K, Bronson R T, Mulligan R C. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 24.Hooper M, Hardy K, Handyside A, Hunter S, Monk M. Nature (London) 1987;326:292–295. doi: 10.1038/326292a0. [DOI] [PubMed] [Google Scholar]
- 25.Koller B H, Kim H-S, Latour A M, Brigman K, Boucher R C, Jr, Scambler P, Wainwright B, Smithies O. Proc Natl Acad Sci USA. 1991;88:10730–10734. doi: 10.1073/pnas.88.23.10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krege J H, Hodgin J B, Hagaman J R, Smithies O. Hypertension. 1995;25:1111–1115. doi: 10.1161/01.hyp.25.5.1111. [DOI] [PubMed] [Google Scholar]
- 27.Kim H-S, Krege J H, Kluckman K D, Hagaman J R, Hodgin J B, Best C F, Jennette J C, Coffman T M, Maeda N, Smithies O. Proc Natl Acad Sci USA. 1995;92:2735–2739. doi: 10.1073/pnas.92.7.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 29.Azrolan N, Breslow J L. J Lipid Res. 1990;31:1141–1146. [PubMed] [Google Scholar]
- 30.Burt D W, Mullins L J, George H, Smith G, Brooks J, Pioli D, Brammar W J. Gene. 1989;84:91–104. doi: 10.1016/0378-1119(89)90143-1. [DOI] [PubMed] [Google Scholar]
- 31.Huang P L, Huang Z, Mashimo H, Bloch D K, Moskowitz M A, Bevan J A, Fishman M C. Nature (London) 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 32.Widdop R E, Gardiner S M, Kemp P A, Bennett T. Br J Pharmacol. 1992;105:653–656. doi: 10.1111/j.1476-5381.1992.tb09034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elsner D, Müntze A, Kromer E P, Riegger G A J. Am J Hypertens. 1992;5:288–291. doi: 10.1093/ajh/5.5.288. [DOI] [PubMed] [Google Scholar]
- 34.Stamler J S, Loh E, Roddy M-A, Currie K E, Creager M A. Circulation. 1994;89:2035–2040. doi: 10.1161/01.cir.89.5.2035. [DOI] [PubMed] [Google Scholar]
- 35.Sigmon D H, Carretero O H, Beierwaltes W H. Am J Physiol. 1992;263:F256–F261. doi: 10.1152/ajprenal.1992.263.2.F256. [DOI] [PubMed] [Google Scholar]
- 36.Salazar F J, Pinilla J M, Romero J C, Quesada T. Hypertension. 1992;20:113–117. doi: 10.1161/01.hyp.20.1.113. [DOI] [PubMed] [Google Scholar]
- 37.Hu L, Manning R D, Jr, Brands M W. Hypertension. 1994;23:185–194. doi: 10.1161/01.hyp.23.2.185. [DOI] [PubMed] [Google Scholar]
- 38.Münter, K. & Hackenthal, E. (1991) J. Hypertens. 9, Suppl. 6, S236–S237. [PubMed]
- 39.Kurtz A, Kaissling B, Busse R, Baier W. J Clin Invest. 1991;88:1147–1154. doi: 10.1172/JCI115415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gardes J, Poux J M, Gonzalez M F, Alhenc-Gelas F, Ménard J. Life Sci. 1992;50:987–993. doi: 10.1016/0024-3205(92)90092-4. [DOI] [PubMed] [Google Scholar]
- 41.John S W M, Krege J H, Oliver P M, Hagaman J R, Hodgin B, Pang S C, Flynn T G, Smithies O. Science. 1995;267:679–681. doi: 10.1126/science.7839143. [DOI] [PubMed] [Google Scholar]
- 42.Kilbourn R G, Jubran A, Gross S S, Griffith O W, Levi R, Adams J, Lodato R F. Biochem Biophys Res Commun. 1990;172:1132–1138. doi: 10.1016/0006-291x(90)91565-a. [DOI] [PubMed] [Google Scholar]
- 43.Goode H F, Howdle P D, Walker B E, Webster N R. Clin Sci. 1995;88:131–133. doi: 10.1042/cs0880131. [DOI] [PubMed] [Google Scholar]
- 44.Szabo C, Southan G J, Thiemermann C. Proc Natl Acad Sci USA. 1994;91:12472–12476. doi: 10.1073/pnas.91.26.12472. [DOI] [PMC free article] [PubMed] [Google Scholar]