

Abstract
SR proteins, named for their multiple arginine/serine (RS) dipeptide repeats, are critical components of the spliceosome, influencing both constitutive and alternative splicing of pre-mRNA. SR protein function is regulated through phosphorylation of their RS domains by multiple kinases, including a family of evolutionarily conserved SR protein-specific kinases (SRPKs). The SRPK family of kinases is unique in that they are capable of phosphorylating repetitive RS domains with remarkable specificity and efficiency. Here, we carried out kinetic experiments specially developed to investigate how SRPK1 phosphorylates the model human SR protein, ASF/SF2. By using the start–trap strategy, we monitored the progress curve for ASF/SF2 phosphorylation in the absence and presence of an inhibitor peptide directed at the active site of SRPK1. ASF/SF2 modification is not altered when the inhibitor peptide (trap) is added with ATP (start). However, when the trap is added first and allowed to incubate for a specific delay time, the decrease in phosphate content of the enzyme–substrate complex follows a simple exponential decline corresponding to the release rate of SRPK1. These data demonstrate that SRPK1 phosphorylates a specific region within the RS domain of ASF/SF2 by using a fully processive catalytic mechanism, in which the splicing factor remains “locked” onto SRPK1 during RS domain modification.
Both the assembly of the spliceosome and the specificity of splice-site selection in mammalian cells require SR proteins, which contain one or two RNA-recognition motifs (RRMs) in the N terminus and a signature domain enriched with arginine/serine dipeptide repeats (RS) in the C terminus (1, 2). The SR proteins are phosphorylated and activated by the SRPK and Clk/Sty families of protein kinases (3–5). This posttranslational modification is required for translocation of the SR protein from the cytoplasm to the nucleus (6, 7) and recruitment of the SR proteins from nuclear speckles (also known as splicing-factor compartments) to nascent transcripts for cotranscriptional splicing (8–10). Phosphorylated SR proteins are believed to facilitate 5′ splice-site recognition through interaction with the RS domain in U1–70 K (a component of the U1 small nuclear ribonucleoprotein) and 3′ splice-site selection by means of the RS domains present in the U2AF heterodimer (11–13). Finally, reversal of the kinase action by protein phosphatases appears to be essential for spliceosome activation and the return of splicing factors to nuclear speckles (5). ASF/SF2 is a prototypical SR protein with two RRM domains for RNA binding and a C-terminal RS domain for mediating protein–protein interactions during spliceosome assembly (see Fig. 1C). A total of 20 serine residues lie in the RS domain and are potential sites for phosphorylation. Mutational studies have verified that phosphorylation occurs exclusively in the RS domain (3, 7, 14).
Fig. 1.

Isolation of an SRPK1ΔS–ASF/SF2 complex. (A) Size-exclusion chromatogram of refolded complex. (B) SDS/12% PAGE of size-exclusion peak. (C) Domain structure of ASF/SF2. The two RRM domains are shown in red, whereas the C-terminal RS domain is shown in green. All the potential serine phosphorylation sites are shown in red.
Although the importance of phosphorylation of SR proteins is well documented for their biological functions in splicing and trafficking in the cell, little is known about the mechanism of phosphorylation. In general, protein kinases are capable of phosphorylating substrates at more than one site, but these modifications typically occur in a “distributive” manner, in which each catalytic event is separated by dissociation and reassociation of the enzyme. These bimolecular steps presumably allow repositioning of the kinase with respect to the new phosphorylation sites in the substrate. Distributive mechanisms are identifiable because of the liberation of free enzyme and partially phosphorylated intermediates after each catalytic event. In one case, the nonreceptor protein tyrosine kinase, Src, phosphorylates numerous sites in the protein substrate Cas without releasing intermediate phosphoproteins (15, 16). The mechanism seems to involve the recognition of newly phosphorylated products by Src's SH2 and SH3 domains, a process that allows repositioning of the enzyme and substrate for processive phosphorylation events.
SR protein phosphorylation represents an interesting paradigm to investigate protein phosphorylation mechanisms. Unlike most phosphorylation sites, the RS dipeptide is limited in terms of information for recognition by most protein kinases, which typically require four to six amino acids (17). Thus, to simultaneously achieve high specificity and high efficiency, it is possible that SRPKs must recognize a specific sequence motif to initiate the kinase reaction and then follow a processive mechanism to catalyze successive phosphorylation in the highly repetitive RS domain. Unlike Src, SRPK1 does not have a separate domain to bind intermediate phosphoproducts. However, modeling for substrate recognition with a newly solved crystal structure of an SRPK in yeast (Sky1p) predicts that the catalytic core of the enzyme requires alternating positively and negatively charged amino acids for substrate recognition (18). Thus, a phosphorylated serine (which is negatively charged) bracketed by positively charged arginines might be a critical signal for phosphorylation of the downstream serine. To test this model, in this article, we determined whether SRPK1 acts as a processive kinase on ASF/SF2.
Processivity has been observed in several enzymes but is most commonly identified with the DNA polymerases (19–24). Some typical methods designed to establish processivity in this enzyme family rely on trapping free polymerase by using agents such as heparin (25), poly(A)·oligo(dT) (26), poly(dI-dC) (27), and activated calf thymus DNA (28). To determine whether SRPK1 dissociates from ASF/SF2 after each phosphorylation event, we modified the start–trap protocol so that it could be applied to protein phosphorylation. By using a peptide inhibitor that binds to the active site of SRPK1 (trap), we showed that the rate and extent of ASF/SF2 polyphosphorylation is not altered by the simultaneous addition of the inhibitor and ATP. In contrast, if the inhibitor is added to a complex of ASF/SF2 and SRPK1 and allowed to incubate, decreases in ASF/SF2 phosphate content are obtained and can be used to assess the release rate of the splicing factor. The data now demonstrate that SRPK1 “locks” onto ASF/SF2 and processively phosphorylates the RS domain.
Materials and Methods
Materials. ATP, 2-(N-morpholino)ethanesulfonic acid (Mes), magnesium chloride, sodium chloride, acetic acid, DE 52 resin, and liquid scintillant were obtained from Fisher Scientific. Polyprep chromatography columns were obtained from BioRad, synthetic substrate and inhibitor peptides were obtained from the USC/Norris Comprehensive Cancer Center Microchemical Core Facility, and [γ-32P]ATP was obtained from NEN.
Expression and Purification of SRPK1ΔS and ASF/SF2. The cDNA encoding SRPK1ΔS was amplified and inserted into pET15b (Novagen), and the recombinant plasmid was then transformed into the BL21(DE3) Escherichia coli strain. The cultures were grown at 37°C in 2 liters of LB broth supplemented with 100 μg/ml ampicillin, and induced with 0.1 mM isopropyl β-d-thiogalactoside at room temperature for 12–16 h. Pelleted cells were lysed by sonication in 150 ml of lysis buffer [50 mM NaCl/20 mM Mes, pH 6.5/20% (vol/vol) glycerol/1 mM PMSF/0.5 ml of protease inhibitor mixture (Sigma)]. The soluble fraction was applied to a 2-ml Ni2+-Sepharose column, and the bound protein was then eluted in two steps: first, with 25 mM imidazole, and second, with 150 mM imidazole. Concentration measurements of SRPK1ΔS were made by using the method of Gill and von Hippel (29). Wild-type ASF/SF2 and a truncated form lacking a portion of the RS domain, ASF/SF2Δ219, were designed and expressed at high levels in the bacterial pellet.
Formation of the SRPK1ΔS–ASF/SF2 Complex. A cell pellet of E. coli overexpressing His-ASF/SF2 is solubilized in denaturing buffer A (6 M urea/500 mM NaCl/20 mM Tris·HCl, pH 7.5/3 mM DTT/1 mM EDTA/0.1 mM PMSF/10% glycerol). A soluble extract is prepared by sonication, followed by high-speed centrifugation to remove cellular debris. The supernatant is loaded onto a Ni2+-Sepharose column equilibrated with buffer A, and pure His-ASF/SF2 is eluted by using 200 mM imidazole. Stoichiometric amounts of pure denatured His-ASF/SF2 and SRPK1ΔS are mixed in 100 ml of buffer A and dialyzed against 2 liters of buffer B (buffer A without urea) for 6 h. Dialysis is repeated two more times, followed by centrifugation to remove trace amounts of precipitate formed during dialysis. After concentration using Amicon ultrafiltration (Millipore), the sample is loaded onto a size-exclusion column (Superdex 200, Amersham Pharmacia). Fractions constituting a dominant peak containing the complex are pooled and subjected to SDS/PAGE analysis. Concentration measurements of the SRPK1ΔS–ASF/SF2 complex were made by using the method of Gill and von Hippel with a 1:1 stoichiometry of enzyme and substrate (29). Analytical size-exclusion chromatography was performed by using a Zorbax GF-250 column (Agilent Technologies, Palo Alto, CA), and apparent molecular weights were determined by using Bio-Rad HPLC standards. A complex containing SRPK1ΔS and ASF/SF2Δ219 was also refolded and purified by using the same protocol.
Column Separation Assay. Steady-state kinetic parameters for SRPK1ΔS with a substrate peptide (YRTRDAPRERSPTR) were determined at 23°C in 50 mM Mes, pH 7.0/10 mM MgCl2/[γ-32P]ATP (600–1,000 cpm·pmol–1). Enzyme, MgCl2, and ATP were preequilibrated for 1 min before initiating the reaction with the addition of substrate. Reactions (20 μl) were quenched with 180 μl of 30% acetic acid, the products were applied to DE 52 columns (3 ml), which were washed with 30% acetic acid (5 ml), and the phosphorylated substrate was collected and its radioactivity was measured. Control experiments were performed to determine the background phosphorylation (i.e., phosphorylation of substrate peptide in the presence of quench).
Autographic Assay for ASF/SF2 Phosphorylation. The phosphorylation of the SRPK1ΔS:ASF/SF2 complex was carried out in 50 mM Mes (pH 7.0)/10 mM free MgCl2/1 mg/ml BSA/[γ-32P]ATP (600–1,000 cpm·pmol–1) at 23°C. Reactions were then quenched with 10 μl of SDS/PAGE loading buffer. A portion of each quenched reaction mixture (20 μl) was separated by using an SDS/12% PAGE gel, and the protein bands corresponding to phosphorylated ASF/SF2 were cut from the dried SDS/PAGE gel and quantitated on the 32P channel in liquid scintillant.
Data Analysis. The progress-curve data were plotted as a ratio of phosphate incorporated to total ASF/SF2 concentration ([32P]/[ASF/SF2]o) as a function of time and were fit to either a single- or double-exponential function. In the latter case, the amplitude of the first phase (α1) represents the fraction of sites phosphorylated in the enzyme–substrate complex. The fraction bound of ASF/SF2 is then calculated from the ratio of α1 and the total amplitude (αtot) (Fraction bound = α1/αtot). The dissociation constant for the SRPK1ΔS–ASF/SF2 complex (Kd), measured by using dilution studies, was determined by plotting the fraction bound as a function of [ASF/SF2]o and fitting the curve to Eq. 1.
Fraction bound =
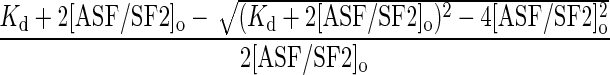 |
[1] |
Eq. 1 represents a quadratic solution for a high-affinity, enzyme–substrate interaction (30) under the special conditions, where [SRPK1ΔS]o = [ASF/SF2]o at all dilutions of the complex.
Results
Formation of a Stable SRPK1–ASF/SF2 Complex for Kinetic Studies. Previous biochemical characterizations indicate that the SRPK family members possess high affinity for RS domain-containing splicing factors (3, 4, 31, 32), making it possible to prepare a stable kinase–substrate complex for the start–trap strategy described below. We prepared recombinant SRPK1 and ASF/SF2 and purified them to homogeneity for the current study. Here we expressed a version of SRPK1, which lacks the insert domain (approximately 250 residues) within the kinase domain. Removal of this insert in the yeast family member, Sky1p, had no effect on catalytic activity (18). Likewise, comparison between His-tagged full-length and the truncated SRPK1 lacking this insert (SRPK1ΔS) revealed similar kinetic properties with both recombinant proteins by using a peptide-based assay (data not shown). Unlike recombinant SRPK1 and SRPK1ΔS, His-tagged ASF/SF2 is not soluble when expressed in E. coli, and refolding from 6 M urea appears rather inefficient (data not shown). To generate an active, unphosphorylated SRPK1–ASF/SF2 complex for kinetic studies, we developed a protocol that uses SRPK1 as a template for the refolding of ASF/SF2. As shown in Fig. 1 A and B, a pure complex of SRPK1ΔS and ASF/SF2 is obtained by using this procedure. No aggregates are apparent in the flowthrough fraction of a sizing column, and the major single peak corresponds to an apparent molecular weight of 75,000 (predicted dimer mass, 78,000 Da). In practice, we found that the refolding protocol worked best with the truncated enzyme form so that all kinetic experiments were performed with the purified SRPK1ΔS–ASF/SF2 complex.
ASF/SF2 Can Be Efficiently and Quantitatively Phosphorylated in the Complex. As shown in Fig. 2, when the SRPK1ΔS–ASF/SF2 complex (0.6 μM) is mixed with [32P]ATP (200 μM), ASF/SF2 readily incorporates phosphates. In this progress curve, the complex is diluted from a concentrated stock (4 μM) into [32P]ATP with no delay time between dilution and reaction initiation. The time course for this modification fits well to a single-exponential progress curve with a forward net rate constant (kf) of 0.80 ± 0.08 min–1 and a total incorporation of 9 ± 0.5 phosphates per ASF/SF2 molecule. This amount is consistent with a report of total phosphates transferred to ASF/SF2 by native SRPK1 (3), indicating that the refolded complex is fully active. To identify which region of the RS domain becomes modified by the enzyme, we generated a complex of SRPK1ΔS and a mutant form of ASF/SF2 lacking sequences beyond residue 219. In progress curves, the deletion mutant (ASF/
Fig. 2.

Progress curve for the phosphorylation of ASF/SF2. SRPK1ΔS–ASF/SF2 is diluted to 0.6 μM in a buffer containing 200 μM [32P]ATP, and phosphate incorporation was measured at various times from 10 s to 45 min by using autoradiography. The data were fit to an exponential function with a forward rate constant (kf) of 0.80 ± 0.08 min–1 and an amplitude of 9 ± 0.5 phosphates.
SF2Δ219) is phosphorylated to the same extent as the wild-type splicing factor (data not shown), indicating that serines between residues 198 and 218 are likely sites of modification within the RS domain. Indeed, a stretch of eight arginine/serine dipeptide repeats within this sequence [(RS)8] may serve as the target sequence for SRPK1 (Fig. 1C).
To determine whether a maximum number of active sites are bound with ATP, a progress curve was measured by using 100 μM [32P]ATP and 0.6 μM complex. Because no change in phosphate content or rate was observed at this lower ATP level (data not shown), the nucleotide concentration in the reaction is saturating. The progress curve for SRPK1ΔS–ASF/SF2 (0.6 μM) was also monitored in the absence and presence of 2.4 and 4.8 μM SRPK1ΔS. Although this increases the apparent stoichiometry of enzyme to substrate from 1:1 to 9:1, no increases in the rate or total phosphate content of ASF/SF2 were observed (data not shown). Progress curves were also measured at a series of SRPK1ΔS–ASF/SF2 concentrations ranging from 0.6 to 4.2 μM, but no significant change in kf or normalized phosphate content was observed within experimental limits if the complex was diluted into [32P]ATP without a delay time (data not shown). Thus, a highly stable, functional complex can be formed over a wide range of protein and nucleotide concentrations.
SRPK1ΔS and ASF/SF2 Form a Dynamic and Functional Kinase–Substrate Complex. To measure the stability of the SRPK1ΔS–ASF/SF2 complex, we monitored 32P incorporation as a function of complex concentration (0.03–4 μM) and time between dilution and addition of 200 μM ATP (dilution time). At high complex concentrations (1–4 μM), the progress curves for ASF/SF2 phosphorylation did not vary as a function of dilution time (up to 15 min) before reaction initiation with ATP (data not shown), indicating that the complex does not dissociate appreciably at or above 1 μM within this time frame. At lower complex concentrations, however, the progress curves become noticeably biphasic as a function of dilution time. As shown in Fig. 3A, the phosphorylation of 0.1 μM ASF/SF2 after a dilution time of 5 min is best fit by a double-exponential function with rate constants that differ by >10-fold (0.8 min–1 for the initial, fast phase, and 0.02 min–1 for the second, slower phase). The biphasic nature of this profile is due to dissociation of the complex into free SRPK1ΔS and ASF/SF2.
Fig. 3.

Complex stability. (A) Progress curves under dissociating conditions. SRPK1ΔS–ASF/SF2 was diluted from 4 to 0.1 μM in a buffer containing 200 μM [32P]ATP, and phosphorylation was monitored by using autoradiography (•). In a separate experiment, the complex was diluted to 0.1 μM and allowed to incubate for 5 min before addition of 200 μM [32P]ATP (○). The data at zero delay time were fit to a single-exponential function with an amplitude of 9.5 ± 0.40 phosphates. The data at the 5-min dilution were fit to a double-exponential function with amplitudes of α1 = 6.1 ± 0.4 and α2 = 3.4 ± 0.4 phosphates. (Inset) Values for α1 at a series of dilution times (0–15 min). The line drawn through the data was obtained from kinetic simulations (see text). (B) Dissociation constant for SRPK1ΔS–ASF/SF2. Progress curves for ASF/SF2 phosphorylation were measured a several complex concentrations. The fraction bound was determined from α1 after 10-min dilution times and plotted against total ASF/SF2 concentration. The data are fit to Eq. 1 to obtain a Kd of 50 ± 25 nM.
To determine that enzyme, substrate, and complex are in dynamic equilibrium, the amplitude of the first phase (α1) was measured as a function of dilution time. As shown in Fig. 3A Inset, α1 declines as a function of time and reaches an equilibrium level after 5 min, implying that free enzyme and substrate can rebind after dissociation. To determine the dissociation constant for SRPK1ΔS–ASF/SF2, the complex was diluted and allowed to reach equilibrium; the amount of enzyme–substrate complex (α1) was then determined by adding [32P]ATP. In Fig. 3B, the fraction of bound ASF/SF2 is plotted as a function of total ASF/SF2 concentration. Fitting the data to Eq. 1 provides a Kd of 50 ± 25 nM. These studies clearly show that the SRPK1ΔS–ASF/SF2 complex, although quite stable, is also dynamic, reflecting the high affinity and functional interaction between the kinase and the substrate. Furthermore, the ability to monitor time-dependent dissociation of the complex (Fig. 3A Inset) provides a means for estimating the association (kon) and dissociation (koff) rate constants for the enzyme and substrate. By using the simulation program kinsim (33), we established values for kon and koff that satisfy the observed dissociation kinetics. The line drawn through the data in Fig. 3A Inset represents the kinetic simulation for a simple dissociation mechanism where kon and koff are set at 11 μM·min–1 and 0.24 min–1, respectively, at an initial complex concentration of 0.1 μM. In general, we found that the experimental data set could be adequately fit by a kon between 5 and 13 μM·min–1 and a koff between 0.18 and 0.30 min–1. These simulated values generate a calculated Kd between 22 and 25 nM, a range that is close to the experimental Kd (50 ± 25 nM).
Development of Peptide Inhibitor for SRPK1. To determine that SRPK1 is a processive enzyme, we designed an efficient trap ligand that can compete with the RS domain of ASF/SF2. We synthesized a 14-residue peptide substrate (YRTRDAPRERSPTR) based on the single phosphorylation site in the SR-related protein, Npl3, and found that it is a good substrate for SRPK1ΔS (Km = 200 ± 20 μM). An inhibitor version of this peptide, AlaPep, was then synthesized by replacing the phosphorylatable serine with alanine (YRTRDAPRERAPTR). AlaPep could not be phosphorylated in the column assay and served as a competitive inhibitor with respect to the substrate peptide (Ki = 30 ± 6 μM).
Start–Trap Experiments Demonstrate the Processivity of SRPK1 in Phosphorylating ASF/SF2. To assess whether SRPK1ΔS phosphorylates ASF/SF2 in succession without dissociating from the RS domain (processive phosphorylation), we designed a start–trap experiment in which ATP (start) and AlaPep (trap) are added simultaneously to the enzyme–substrate complex (Fig. 4A). If the enzyme dissociates from the substrate after each phosphorylation event (pathway 2, distributive phosphorylation), AlaPep will trap the free enzyme, thereby stopping the phosphorylation reaction after one round of phosphorylation. Alternatively, if SRPK1ΔS phosphorylates all available serines without dissociating, the inhibitor will not influence the rate or extent of phosphate incorporation into ASF/SF2 (pathway 1, processive phosphorylation). As shown in Fig. 4B, the presence of AlaPep (20 mM) has no effect on the rate or total incorporation of phosphate, an observation that strongly supports the processive pathway.
Fig. 4.

Start–trap experiment. (A) Experimental design. SRPK1ΔS–ASF/SF2 is mixed simultaneously with [32P]ATP (start) and AlaPep (trap). In this experimental scheme, the enzyme and AlaPep are shown in blue and red, respectively. The two RRMs of ASF/SF2 are shown in green. The unphosphorylated RS domain is designated with open circles. One phosphorylated serine is shown with a filled circle. Pathways 1 and 2 describe processive and distributive mechanisms, respectively. (B) Progress curves with and without trap. SRPK1ΔS–ASF/SF2 (1 μM) is mixed with 200 μM[32P]ATP, and time-dependent phosphorylation is monitored by using the autoradiographic assay in the absence (○) and presence (□) of 20 mM AlaPep. The data are fit to a single-exponential function with a common rate constant of 0.80 ± 0.10 min –1 and amplitude of 9.0 ± 0.5 phosphates.
As a control for the start–trap experiment, we designed the following trap–delay–start experiment to evaluate the ability of the trapping ligand to compete effectively with ASF/SF2 for the active site of SRPK1ΔS. In this experiment, the preformed complex is mixed with AlaPep (trap phase) and allowed to equilibrate for a specified time (delay phase). The delay phase comprises the time needed to dissociate SRPK1ΔS from the RS domain before the concentration of remaining active complex can be assayed in the start phase. In this experiment, SRPK1ΔS–ASF/SF2 (1 μM) is first preequilibrated with the peptide inhibitor (20 mM) for various delay times (tdelay) before the reaction is started with [32P]ATP (200 μM). The progress curves at each delay time were best fit by single-exponential functions for a 60-min reaction time (data not shown). The observed phosphate incorporation during this period (α1) declines as a function of tdelay (Fig. 5) and is fit to a rate constant of 0.30 ± 0.05 min–1. This value is very close to the simulated off-rate of the enzyme and substrate calculated from the dilution studies in Fig. 3A (0.24 min–1), suggesting that the release rate in the trap–delay–start experiments represents a bimolecular process. In control experiments, the addition of a buffer blank in place of trap led to no appreciable decline in ASF/SF2 phosphate content, implying that the observed changes in Fig. 5 are not due to time-dependent complex inactivation (data not shown). Also, halving the AlaPep concentration did not alter the displacement curve in Fig. 5 (data not shown), suggesting that the peptide concentration is high enough to trap all of the free enzyme monomers. Taken together, the data from the start–trap and trap–delay–start experiments provide the unequivocal evidence that SRPK1 acts as a processive kinase in phosphorylating the SR protein ASF/SF2.
Fig. 5.

Trap–delay–start experiment. SRPK1ΔS–ASF/SF2 complex (1 μM) is mixed with AlaPep (20 mM) and allowed to equilibrate for various delay times (0–12 min) before 200 μM[32P]ATP is added (start phase). The amplitude of the first kinetic phase (α1) in the progress curves is plotted as a function of the delay time and then fit to a single-exponential function with a rate constant of 0.30 ± 0.02 min–1.
Discussion
Although phosphorylated SR proteins are required for initiating spliceosome assembly in multiple steps (12, 34), little is known about where and how they are phosphorylated. To address this issue we purified a stable and active SRPK1ΔS in complex with its natural substrate, ASF/SF2 (Fig. 1). Using autoradiography, we showed that ASF/SF2 is phosphorylated by SRPK1ΔS at ≈9 sites (Fig. 2). Because the observed number of phosphorylation sites is not affected by total ATP concentration, the inability to modify all serines in the RS domain is not the result of an unfavorable equilibrium in the active site of the enzyme. Thus, although the RS domain contains 20 serines, only one-half of these sites are phosphorylated by SRPK1 in vitro. To localize the phosphorylated region, a C-terminal deletion mutant (missing residues 219–248) was analyzed. Because this mutant protein incorporates phosphate to the same extent as wild type, the phosphorylatable region thus appears to be confined within the stretch of eight serines in the N-terminal portion of the RS domain. This result explains why the Drosophila ASF/SF2 homologue could not be phosphorylated by SRPK1 because the RS repeats in this region are replaced by a glycine tract (9).
Specificity of the SR Protein–Protein Kinase Interaction. Using dilution studies, we showed that SRPK1ΔS and ASF/SF2 form a highly stable complex that is in dynamic equilibrium (Fig. 3). Because these studies were performed at a constant 1:1 stoichiometry of enzyme and substrate, we wondered whether additional enzyme monomers could associate with the RS domain at high SRPK1ΔS concentrations. Formation of higher order complexes might accelerate the rate, further increase the phosphate content of ASF/SF2, or alter site selection within the RS domain. None of these had happened because addition of a high level of SRPK1ΔS had little effect on the kinetic profile. Thus, despite the presence of other additional RS repeats, which can be potential targets for phosphorylation, SRPK1 does not appear to initiate phosphorylation at those additional sites even at high stoichiometries. This observation is consistent with the possibility that ASF/SF2 only offers a defined distal structural element(s) that guides only a single SRPK1 to one specific initiation site.
Developing a Simple Protocol for Establishing Processivity in Protein Kinases. Most protein kinases bind a substrate protein, delivering a single phosphate group with ATP as a phosphate donor. Recently, it has been demonstrated that the nonreceptor protein tyrosine kinase Src catalyzes phosphorylation of the protein substrate Cas at numerous sites by using a fully processive kinetic mechanism (16). To define this mechanism, large amounts of free protein substrate were required to establish an effective competition between unphosphorylated and intermediate phosphorylated forms of Cas. A similar strategy cannot be exploited to study SRPK1 by using ASF/SF2, because this recombinant protein substrate will form aggregates at high concentrations. The application of the start–trap experiment in this report removes the requirement for a large amount of protein substrate. This procedure may be generally useful for other protein kinase–protein substrate systems, in particular, when components of the system cannot be expressed at high, soluble levels, a common problem facing enzymological studies on this enzyme family.
ASF/SF2 Is Locked onto SRPK1ΔS. Enzymes that catalyze processive reactions are expected to possess forward catalytic rate constants (kf) that exceed the dissociation rate constant of the enzyme–substrate complex (koff) (24). We measured the dissociation rate constant for SRPK1ΔS by using a trap–delay–start experiment to estimate the commitment of ASF/SF2 toward processive phosphorylation (Fig. 5). The delay time (tdelay) between addition of AlaPep (trap) and ATP (start) defines a period where the peptide can displace the RS domain from the enzyme's active site. Once ATP is added, the reaction proceeds processively so that the time required for measurement of the phosphate content does not provide an additional opportunity for RS domain dissociation and α1 then reflects an estimate of the amount of bound SRPK1ΔS. By using this technique, the measured off-rate (0.30 min–1) was found to be 3-fold lower than kf. Although this satisfies the relationship kf > koff, the level of processivity observed in the start–trap experiment is large compared with the ratio of kf/koff. We believe that this ratio represents a lower limit on the commitment factor for enzyme and substrate. In a previous study, we showed that the yeast SR kinase, Sky1p, modifies the protein substrate, Npl3, in the active site at a rate constant of 40 s–1, a value 80-fold larger than the turnover number (35). If SRPK1 utilizes rapid phosphate transfer, this step could provide a large commitment factor for processive ASF/SF2 phosphorylation. Because phosphorylated substrate is not released from the enzyme at intermediate stages, it is unclear what step or steps then control kf. It is possible that translocation or associated conformational changes could provide the slow steps in this reaction.
Model for Processivity in SRPK1. Using a start–trap experiment, we demonstrated that a large stretch of serines in the RS domain of ASF/SF2 are phosphorylated processively. This observation now raises the questions of how phosphorylation is started, and then what is the molecular basis for consecutive phosphorylation. With regard to the initiation of processive phosphorylation, we predict that sequences upstream from the RS domain, perhaps in the RRM domains, may guide the initial recognition of the substrate by the kinase, thereby locking SRPK1 onto the RS domain. However, processivity may also require local sequence recognition. As described in the introduction, the x-ray structure of Sky1p suggests that two conserved arginines in the kinase recognize a negatively charged amino acid in the P-2 position of a peptide substrate modeled in the active site (-ERS-) (18). In general, the negatively charged amino acid may be required for initiation of processive phosphorylation. For ASF/SF2, once a serine is phosphorylated in the RS repeat, the negatively charged phosphoserine may then serve as the recognition signal for phosphorylation of the downstream serines.
Biological Implication of Processive Phosphorylation of the RS Domain. Processive phosphorylation of SR proteins offers distinctive rate advantages over the more common distributive process. For example, if ASF/SF2 dissociates after each round of phosphorylation at a rate of 0.3 min–1 (Fig. 5B), it is anticipated that the overall rate for RS domain phosphorylation would be ≈25-fold slower than the empirical value of 0.8 min–1 (Fig. 2).
Thus, the incorporation of processivity lowers the mean half-life for ASF/SF2 activation from just over 20 min to 50 s. Given the overall complexity and precise requirements for phosphorylation and dephosphorylation at specific stages of spliceosome assembly and activation (36–39), the timing of SR protein activation could play an important role in splicing. How ASF/SF2 stays “locked” onto the RS domain may also play an important role in splicing. For example, the kinase may function as a molecular switch to determine the timing of RS domain-mediated protein–protein interactions during spliceosome assembly and/or RS domain dephosphorylation during spliceosome activation. These ideas may be tested by using mutant SR proteins that are preserved for their ability to be phosphorylated by other kinases, but are no longer capable of efficiently interacting with the SRPK family of kinases.
Acknowledgments
We thank Dr. Jack Kyte for providing valuable insights into the treatment of the kinetic data and careful reading of the manuscript. This work was supported by National Institutes of Health Grant GM 68168. B.E.A. was supported by National Institutes of Health Training Grant GM 07752.
Abbreviations: AlaPep, inhibitor peptide sequence YRTRDAPRERAPTR; ASF/SF2, human alternative splicing factor; ASF/SF2Δ219, mutant ASF/SF2 lacking residues beyond 219; RS domain, domain rich in arginine/serine repeats; RRM, RNA-recognition motif; SRPK, SR-specific protein kinase; SRPK1ΔS, SRPK1 lacking the domain insert.
References
- 1.Fu, X. D. (1995) RNA 1 663–680. [PMC free article] [PubMed] [Google Scholar]
- 2.Zahler, A. M., Lane, W. S., Stolk, J. A. & Roth, M. B. (1992) Genes Dev. 6 837–847. [DOI] [PubMed] [Google Scholar]
- 3.Gui, J. F., Tronchere, H., Chandler, S. D. & Fu, X. D. (1994) Proc. Natl. Acad. Sci. USA 91 10824–10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang, H. Y., Lin, W., Dyck, J. A., Yeakley, J. M., Songyang, Z., Cantley, L. C. & Fu, X. D. (1998) J. Cell Biol. 140 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stojdl, D. F. & Bell, J. C. (1999) Biochem. Cell Biol. 77 293–298. [PubMed] [Google Scholar]
- 6.Lai, M. C., Lin, R. I. & Tarn, W. Y. (2001) Proc. Natl. Acad. Sci. USA 98 10154–10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koizumi, J., Okamoto, Y., Onogi, H., Mayeda, A., Krainer, A. R. & Hagiwara, M. (1999) J. Biol. Chem. 274 11125–11131. [DOI] [PubMed] [Google Scholar]
- 8.Duncan, P. I., Stojdl, D. F., Marius, R. M., Scheit, K. H. & Bell, J. C. (1998) Exp. Cell Res. 241 300–308. [DOI] [PubMed] [Google Scholar]
- 9.Colwill, K., Pawson, T., Andrews, B., Prasad, J., Manley, J. L., Bell, J. C. & Duncan, P. I. (1996) EMBO J. 15 265–275. [PMC free article] [PubMed] [Google Scholar]
- 10.Gui, J. F., Lane, W. S. & Fu, X. D. (1994) Nature 369 678–682. [DOI] [PubMed] [Google Scholar]
- 11.Xiao, S. H. & Manley, J. L. (1998) EMBO J. 17 6359–6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao, S. H. & Manley, J. L. (1997) Genes Dev. 11 334–344. [DOI] [PubMed] [Google Scholar]
- 13.Jamison, S. F., Pasman, Z., Wang, J., Will, C., Luhrmann, R., Manley, J. L. & Garcia-Blanco, M. A. (1995) Nucleic Acids Res. 23 3260–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Misteli, T., Caceres, J. F., Clement, J. Q., Krainer, A. R., Wilkinson, M. F. & Spector, D. L. (1998) J. Cell Biol. 143 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mayer, B. J., Hirai, H. & Sakai, R. (1995) Curr. Biol. 5 296–305. [DOI] [PubMed] [Google Scholar]
- 16.Pellicena, P. & Miller, W. T. (2001) J. Biol. Chem. 276 28190–28196. [DOI] [PubMed] [Google Scholar]
- 17.Pearson, R. B. & Kemp, B. E. (1991) Methods Enzymol. 200 62–81. [DOI] [PubMed] [Google Scholar]
- 18.Nolen, B., Yun, C. Y., Wong, C. F., McCammon, J. A., Fu, X. D. & Ghosh, G. (2001) Nat. Struct. Biol. 8 176–183. [DOI] [PubMed] [Google Scholar]
- 19.Fairfield, F. R., Newport, J. W., Dolejsi, M. K. & von Hippel, P. H. (1983) J. Biomol. Struct. Dyn. 1 715–727. [DOI] [PubMed] [Google Scholar]
- 20.McClure, W. R. & Chow, Y. (1980) Methods Enzymol. 64 277–297. [DOI] [PubMed] [Google Scholar]
- 21.Kuriyan, J. & O'Donnell, M. (1993) J. Mol. Biol. 234 915–925. [DOI] [PubMed] [Google Scholar]
- 22.Von Hippel, P. H., Fairfield, F. R. & Dolejsi, M. K. (1994) Ann. N.Y. Acad. Sci. 726 118–131. [DOI] [PubMed] [Google Scholar]
- 23.Trakselis, M. A. & Benkovic, S. J. (2001) Structure (London) 9 999–1004. [DOI] [PubMed] [Google Scholar]
- 24.Bambara, R. A., Fay, P. J. & Mallaber, L. M. (1995) Methods Enzymol. 262 270–280. [DOI] [PubMed] [Google Scholar]
- 25.Gopalakrishnan, V., Peliska, J. A. & Benkovic, S. J. (1992) Proc. Natl. Acad. Sci. USA 89 10763–10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeStefano, J. J., Buiser, R. G., Mallaber, L. M., Myers, T. W., Bambara, R. A. & Fay, P. J. (1991) J. Biol. Chem. 266 7423–7431. [PubMed] [Google Scholar]
- 27.Huber, H. E., McCoy, J. M., Seehra, J. S. & Richardson, C. C. (1989) J. Biol. Chem. 264 4669–4678. [PubMed] [Google Scholar]
- 28.Joyce, C. M. (1989) J. Biol. Chem. 264 10858–10866. [PubMed] [Google Scholar]
- 29.Gill, S. C. & von Hippel, P. H. (1989) Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- 30.Taira, K. & Benkovic, S. J. (1988) J. Med. Chem. 31 129–137. [DOI] [PubMed] [Google Scholar]
- 31.Yun, C. Y. & Fu, X. D. (2000) J. Cell Biol. 150 707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mayeda, A., Screaton, G. R., Chandler, S. D., Fu, X. D. & Krainer, A. R. (1999) Mol. Cell. Biol. 19 1853–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barshop, B. A., Wrenn, R. F. & Frieden, C. (1983) Anal. Biochem. 130 134–145. [DOI] [PubMed] [Google Scholar]
- 34.Cao, W., Jamison, S. F. & Garcia-Blanco, M. A. (1997) RNA 3 1456–1467. [PMC free article] [PubMed] [Google Scholar]
- 35.Aubol, B. E., Nolen, B., Vu, D., Ghosh, G. & Adams, J. A. (2002) Biochemistry 41 10002–10009. [DOI] [PubMed] [Google Scholar]
- 36.Hastings, M. L. & Krainer, A. R. (2001) Curr. Opin. Cell Biol. 13 302–309. [DOI] [PubMed] [Google Scholar]
- 37.Philips, A. V. & Cooper, T. A. (2000) Cell. Mol. Life Sci. 57 235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Misteli, T. (2000) J. Cell Sci. 113 1841–1849. [DOI] [PubMed] [Google Scholar]
- 39.Graveley, B. R. (2000) RNA 6 1197–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]