

Abstract
The objective of this study was to clarify the relative roles of medial versus luminal factors in the induction of thickening of the arterial intima after balloon angioplasty injury. Platelet-derived growth factor (PDGF) and thrombin, both associated with thrombosis, and basic fibroblast growth factor (bFGF), stored in the arterial wall, have been implicated in this process. To unequivocally isolate the media from luminally derived factors, we used a 20-μm thick hydrogel barrier that adhered firmly to the arterial wall to block thrombus deposition after balloon-induced injury of the carotid artery of the rat. Thrombosis, bFGF mobilization, medial repopulation, and intimal thickening were measured. Blockade of postinjury arterial contact with blood prevented thrombosis and dramatically inhibited both intimal thickening and endogenous bFGF mobilization. By blocking blood contact on the two time scales of thrombosis and of intimal thickening, and by using local protein release to probe, by reconstitution, the individual roles of PDGF-BB and thrombin, we were able to conclude that a luminally derived factor other than PDGF or thrombin is required for the initiation of cellular events leading to intimal thickening after balloon injury in the rat. We further conclude that a luminally derived factor is required for mobilization of medial bFGF.
Complications of arterial wound healing greatly limit the efficacy of procedures to re-open obstructed arteries. Intimal thickening, which involves the migration and proliferation of smooth muscle cells (SMCs) in the arterial wall and the overproduction of matrix proteins (1), may contribute to these complications. We and others have previously reported the use of thin intravascular hydrogel barriers for the prevention of thrombosis in the rabbit after balloon injury (2) and in the pig after stent deployment (3, 4). The biocompatible barriers, formed via in situ interfacial photopolymerization of a polyethylene glycol (PEG)-based precursor, adhere to the vessel wall and block deposition of blood cells after injury. We sought to use this ability to unequivocally isolate the arterial wall from blood after injury to investigate the role of thrombus-derived factors, such as platelet-derived growth factor (PDGF) and thrombin, in initiating the cascade of events that leads to intimal thickening after arterial injury. This was accomplished using barrier materials that remained on the artery for either the time scale of thrombosis or of intimal thickening, and also using perivascular materials to reconstitute platelet-derived factors by local release.
Thrombus adherent to the arterial intima after injury may provide multiple signals for intimal thickening. PDGF is released from the α-granules of platelets upon activation and is a SMC chemotactic factor and mitogen. PDGF has been shown to induce intimal thickening after balloon injury in the rat (5), primarily by induction of SMC migration and matrix elaboration (6). Thrombin, generated by the prothrombinase complex on activated platelets, is a SMC mitogen and stimulates SMC matrix elaboration (7, 8). Administration of a thrombin inhibitor has been shown to reduce intimal thickening after balloon injury in the rabbit (9). The platelet–fibrin thrombus may also serve as a physical matrix for SMC migration (10).
Factors intrinsic to the arterial media may also stimulate intimal thickening. Basic fibroblast growth factor (bFGF) is present in the normal arterial media, both intracellularly and bound in inactive form to heparan sulfate proteoglycan in the extracellular matrix (11, 12). bFGF has been shown to promote SMC chemotaxis and proliferation. Local delivery of bFGF after balloon injury in the rat greatly increased intimal thickening and SMC proliferation (13). Treatment with bFGF neutralizing antibodies after arterial injury decreased SMC proliferation by ≈80%, yet did not significantly affect intimal thickening (14).
In spite of many important insights into the biological responses induced by individual factors involved in arterial healing, the relationship between these factors following injury remains unclear. One reason for this is the difficulty in separating blood-derived factors from vessel wall-derived factors following injury. We now report an investigation of the relative roles of luminal versus medial factors in arterial healing using a novel approach to physically separate the vessel wall from the lumen.
MATERIALS AND METHODS
Rat Balloon Injury Model.
A balloon-induced deendothelialization and stretch injury to the carotid artery of the rat was used as an experimental model somewhat analogous to balloon angioplasty, although the injury created is more severe than that in the clinical situation. In addition, the adventitia was removed from the arterial wall in this model; while this may impact arterial healing, the rat has minimal vasa vasorum and thus the effect would be expected to be small. Male Sprague Dawley rats (300–400 g) were anesthetized i.p. with pentobarbital (50 mg/kg). The left carotid artery was surgically exposed, and microsurgical clamps were applied to the internal, external, and common carotid arteries to isolate a zone in the common carotid artery. The adventitia was dissected from the zone, and blood was removed by rinsing with Hepes-buffered saline (HBS; pH 7.4, 10 mM). A balloon injury was subsequently created by passing an inflated 2F Fogarty catheter through the common carotid artery zone three times. Animals were randomly assigned to treatment and control groups after injury. Immediately after injury but before reexposure to blood flow, the appropriate treatment was applied as described below. Control animals received no treatment. Additional control groups received application of the hydrogel barrier without balloon injury to determine if the photopolymerization process or the prolonged presence of the hydrogel barrier caused morphological changes in the vessel wall; blood was rinsed from the carotid arteries prior to hydrogel treatment. Rats were killed by CO2 asphyxiation at the predetermined time points, and the carotid arteries were rinsed with HBS, fixed with 10% buffered formalin under perfusion, and then explanted and prepared for histological analysis. All animal experimentation was approved by the University of Texas Animal Care and Use Committee.
Interfacial Photopolymerization.
The hydrogel barrier was formed on the balloon-injured arterial wall of the rat via interfacial photopolymerization (2, 15). Following balloon injury, a PE-10 polyethylene tube was inserted as a catheter through the external carotid artery to the common carotid artery to introduce fluids to the artery. The carotid zone was flushed with a tissue-adsorbing photoinitiator, eosin Y (1 mM in HBS), for 30 sec. Excess eosin Y was rinsed with HBS, and the region was filled with a solution of PEG diacrylate (PEG-DA; nondegradable) or PEG-co-polylactic acid diacrylate (PEG-L-DA; degradable) (both 23%) with cocatalysts triethanolamine (100 mM) and N-vinylpyrrolidone (1500 ppm) in HBS and illuminated with a xenon arc lamp [Optomed, Austin, TX; 400–600 nm, 35 mW/cm (2), 30 sec for PEG-DA or 10 sec for PEG-L-DA]. All solutions were sterilized by filtration (0.2 μm). The interfacial nature of the photopolymerization process, with the initiator adsorbed to the arterial wall, constrained formation of the hydrogel to the intimal surface. The catheter was removed, and the external carotid artery was ligated with a 5-0 silk suture. In control experiments, the hydrogel barrier was applied to noninjured arteries.
Determination of Hydrogel Thickness and Degradation Kinetics.
Fluorescent beads (1 μm diameter, Polysciences) were mixed into the precursor formulation and became entrapped within the resulting gel, aiding in visualization as described previously (2). The hydrogel barrier was formed by interfacial photopolymerization of PEG-DA or PEG-L-DA as described above in noninjured animals. Animals were killed at predetermined time points by CO2 asphyxiation, and the left carotid artery was immediately explanted and rinsed. The vessels were cryosectioned and visualized by fluorescence microscopy. Digital image analysis (Hamamatsu, Middlesex, NJ) was used to measure the thickness of the hydrogel barrier at eight equally spaced angles around the artery.
Local Release of PDGF and Thrombin.
PEG-L-DA (23%) was dissolved in solutions of PDGF (BB form, 620 μg/ml, Sigma), thrombin (9.3 units/ml, Sigma), or HBS (control), each of which also contained 0.1 mM eosin Y, 100 mM triethanolamine, and 1500 ppm N-vinylpyrrolidone. All solutions were sterilized by filtration (0.2 μm). A balloon injury was created as described above, after which assignment to a treatment grouping was made. If appropriate for the given treatment group, a PEG-DA barrier was applied by interfacial photopolymerization before reexposure to blood flow, and 100 μl of the appropriate PEG-L-DA solution was applied to the outside of the common carotid artery. The liquid precursor was converted to a hydrogel by exposure to a xenon arc lamp [20 mW/cm (2), 30-sec exposure]. Controls consisted of animals lacking the intimal PEG-DA barrier or lacking PDGF-BB or thrombin in the perivascular PEG-L-DA hydrogel. At 14 days, the rats were killed, and the arteries were fixed and explanted as described above and then prepared for histological analysis. The quantities of PDGF-BB and thrombin that were delivered were based on approximately twice the amount contained within a thrombus 50 μm thick (15, 16).
Histological Analysis.
Histological staining was used to quantify thrombosis, intimal thickening, total cells in the media, and inflammatory cells in the media. To assess the extent of thrombosis, sections were stained with hematoxylin and eosin, and the thickness of the thrombus was measured at eight equally spaced angles around the artery. To assess the extent of intimal thickening, sections were stained with van Gieson’s elastin stain; the cross-sectional areas of the intima (all tissue internal to the internal elastic lamina) and the media were measured by digital image analysis (Hamamatsu). To determine the total number of cells in the media, sections were stained with hematoxylin and eosin, and all cell nuclei in the media were counted. To determine the number of inflammatory cells in the media, sections were stained with Giemsa stain, and Giemsa-positive cells were counted; any preferential localization of Giemsa-positive cells was noted.
Immunohistochemical staining was used to identify medial SMCs. Sections were deparaffinized, rehydrated, and partially digested with trypsin (0.1 mg/ml, 10 min). Sections were incubated in 10 mg/ml BSA for 1 hr and then in mouse anti-SMC actin IgG (Dako, 1:100) for 6 hr. After thorough rinsing, the sections were incubated in horseradish peroxidase-conjugated goat anti-mouse IgG (Dako, 1:100) for 2 hr and then developed with diaminobenzidine (Sigma). A cell nucleus (by hematoxylin counterstain) not within one nuclear diameter of SMC actin-positive staining was judged to be other than a SMC.
bFGF Depletion.
Arteries were balloon injured and treated with the PEG-DA hydrogel barrier as described above. On day 4, the rats were killed by CO2 asphyxiation, and the left (injured) and right (not injured or treated) carotid arteries were prepared for assay of bFGF exactly as described (17). Blood was thoroughly rinsed from the segments, and the tissue was frozen in liquid nitrogen and ground with mortar and pestle to form a fine powder. This powder was resuspended in 1% SDS containing 0.2 mg/ml EDTA, 1 μM phenylmethylsulfonyl fluoride, and 10 μg/ml leupeptin (each from Sigma). Particulate matter was removed by centrifuging at 10,000 × g for 5 min. Total protein concentration in each sample was determined by BCA assay (Pierce). bFGF levels (relative to total protein) were measured by ELISA using a mouse anti-human bFGF IgG (which cross reacts with rat bFGF, Sigma), a HRP-conjugated goat anti-mouse IgG (Sigma), and 3,3′,5,5′-tetramethylbenzidine as the horseradish peroxidase substrate (Pierce).
Precursor Synthesis.
PEG-L-DA was synthesized as described previously (2). Briefly, PEG (10,000 Da) was dried by azeotropic distillation in toluene. A 7-fold molar excess (based on end-groups) of DL-lactide and equimolar stannous octoate were added to the polymer solution. The mixture was refluxed under argon for 16 hr and cooled to room temperature; PEG-L was precipitated with hexane, filtered, washed, and dried. The PEG-L was then dissolved in tetrahydrofuran and reacted with a 5-fold molar excess of acryloyl chloride and a 5-fold molar excess of triethylamine for 24 hr under argon at room temperature. The mixture was filtered, and the PEG-L-DA was precipitated with cold ether, filtered, washed, and dried. PEG-L-DA was stored at −20°C under argon until use.
PEG-DA was synthesized as described (18). Briefly, PEG (10,000 Da) was dried by azeotropic distillation, dissolved in tetrahydrofuran and reacted with a 5-fold molar excess of acryloyl chloride and a 5-fold molar excess of triethyl amine for 24 hr under argon. The mixture was filtered, and the PEG-DA was precipitated with cold ether, filtered, washed, and dried.
RESULTS
Stability of the Hydrogel Barrier.
The stability of the PEG-DA hydrogel barrier (nondegradable) was examined in noninjured rat carotid arteries after cryosectioning. The thickness of the barrier can be controlled via the duration of illumination (2): a thickness of 19 ± 7 μm (mean ± SEM, n = 4) was obtained by illumination for 30 sec at 35 mW/cm (2) with PEG-DA. The thickness of the PEG-DA hydrogel barrier did not change significantly over 28 d in vivo (19 ± 7 μm prior to blood flow versus 16 ± 6 μm at day 28, mean ± SEM, n = 4 per group, P > 0.5 by the Mann–Whitney U test).
Degradation of the PEG-L-DA hydrogel barrier was assessed by monitoring the disappearance of the barrier over time. The initial thickness of the PEG-L-DA barrier was 16 ± 4 μm (mean ± SEM, n = 4 per group). At 6 hr, the thickness was reduced to 13 ± 2 μm, and at 12 hr the hydrogel formed a continuous barrier with a thickness of 8 ± 4 μm. At 24 hr, the hydrogel barrier could no longer be detected on the arterial wall.
Thrombosis, Intimal Thickening, and Medial Repopulation.
Blood contact was prohibited after balloon injury by applying the hydrogel barrier prior to reexposure to blood flow. This process altered both thrombosis and intimal thickening. Thrombosis was completely prevented by application of the nondegradable hydrogel barrier while control vessels consistently displayed mural thrombosis with an average thickness of 49 ± 23 μm (P < 0.005) at 2 hr (Fig. 1 A and B). In vessels treated with the degradable PEG-L-DA barrier, present on the arterial wall for ≈24 hr, thrombosis was completely prevented as measured 2 hr following injury. The presence and time course of thrombosis following degradation of the PEG-L-DA barrier was not examined. The long-term healing outcome of intimal thickening was histologically assessed at 14 and 28 days (Table 1; Fig. 1 C and D). Blockade of blood contact with the nondegradable PEG-DA hydrogel barrier reduced intimal thickening by 95% and 91% relative to untreated controls at 14 and 28 days. Treatment with the degradable PEG-L-DA barrier reduced intimal thickening by 90% at 14 days. There was not a statistically significant difference in intimal thickening for groups treated with degradable versus nondegradable barriers (P > 0.5).
Figure 1.
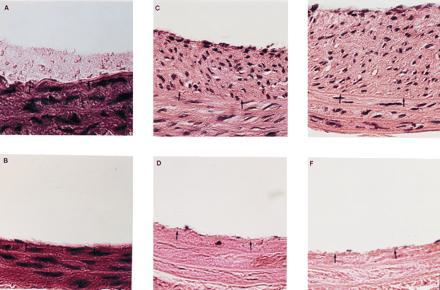
Histology of rat carotid arteries after balloon injury and various treatments as described below. All barriers were nondegradable and remained adherent to the artery wall for the duration of the experiment. The hydrogel barrier cannot be observed in these sections due to the dehydration process. Staining was by hematoxylin and eosin. Black arrows denote the location of the internal elastic lamina. (Bar = 50 μm.) (A) Control vessel 2 hr after injury. Platelet deposition can be clearly noted above the two arrowheads. (B) Hydrogel-treated vessel 2 hr after injury. No platelet deposition could be noted by light microscopy. (C) Control vessel 28 days after injury. The formation of a fibrocellular neointima is clearly evident above the two arrowheads. (D) Hydrogel-treated vessel 28 days after injury. Isolation from blood contact prevented neointima formation and slowed repopulation of the media. (E) Vessel 14 days after injury, with local delivery of PDGF but without blockade of blood contact. Under these conditions, local delivery of PDGF enhanced intimal thickening. (F) Vessel 14 days after injury, with local delivery of PDGF and with barrier blockade of blood contact. Without blood contact, local delivery of PDGF had no effect upon intimal thickening or medial repopulation.
Table 1.
Intimal thickening of control and hydrogel-treated rat carotid arteries 14 and 28 days after balloon-induced injury
| Group (n)* | I/M† | Intimal area, mm2 | Significance relative to control‡ |
|---|---|---|---|
| Control, 14 days (7) | 0.92 ± 0.15 | 0.22 ± 0.05 | |
| PEG-L-DA, 14 days (7) | 0.09 ± 0.03 | 0.02 ± 0.01 | P < 0.001 |
| PEG-DA, 14 days (7) | 0.05 ± 0.02 | 0.01 ± 0.01 | P < 0.001 |
| Control, 28 days (10) | 1.13 ± 0.16 | 0.22 ± 0.03 | |
| PEG-DA, 28 days (10) | 0.10 ± 0.03 | 0.03 ± 0.01 | P < 0.001 |
Values are mean ± SEM.
PEG-L-DA, degradable hydrogel barrier that blocked blood contact with the artery wall for approximately 24 hr; PEG-DA, nondegradable hydrogel barrier that blocked blood contact for the duration of the experiment. Controls were injured but not treated with a hydrogel barrier. n, Number of animals per group.
Ratio of the cross sectional areas of the intima and media.
By the Mann–Whitney U test.
Repopulation of the media with SMCs after injury, presumably due to migration of SMCs from uninjured portions of the artery, was slowed by prolonged blockade of blood contact (Table 2). At 2 days, all injured arteries were devoid of cell nuclei within the media. In the control group, with normal exposure to blood after injury, medial repopulation was completed by day 14 (460 medial cells per cross-section versus 459 in control injured arteries at 28 days, and versus 516 in normal arteries without injury or treatment, P > 0.2), while in groups with prolonged blockade of blood contact, medial repopulation was still incomplete at day 28 (312 medial cells per cross-section, P < 0.05). This delay in medial repopulation was not observed in the group treated with the degradable PEG-L-DA barrier.
Table 2.
Medial repopulation of control and hydrogel-treated arteries after balloon-induced injury
| Group (n) | Number of medial cells/cross section* | Significance relative to control† |
|---|---|---|
| Control, 2 days (4) | 0 ± 0 | |
| PEG-DA, 2 days (4) | 0 ± 0 | P = 1.0 |
| Control, 14 days (7) | 460 ± 43‡ | |
| PEG-L-DA, 14 days (7) | 388 ± 40§ | P > 0.2 |
| PEG-DA, 14 days (7) | 312 ± 39¶ | P < 0.05 |
| Control, 28 days (10) | 459 ± 30 | |
| PEG-DA, 28 days (10) | 336 ± 52 | P < 0.05 |
| PEG-DA, 28 days no injury (4) | 500 ± 39 | P > 0.1 |
| No injury or treatment (4) | 516 ± 45 |
Values are mean ± SEM.
Measured by counting cell nuclei in histological sections stained with hematoxylin and eosin.
By the Mann–Whitney U test.
6.7 ± 4.3 cells per cross-section were non-SMC, and 0.86 ± 0.90 were inflammatory.
6.4 ± 4.7 cells per cross-section were non-SMC, and 0.43 ± 0.53 were inflammatory.
5.3 ± 3.7 cells per cross-section were non-SMC, and 0.71 ± 1.11 were inflammatory.
The identity of the cells participating in repopulation of the media at 14 days was examined histologically. Virtually all cells in the media were positive for SMC actin with no statistically significant differences in the number of cells that were SMC actin-negative between groups. Very few inflammatory cells were noted in any of the groups, with no statistically significant differences between groups. The number of SMC actin-negative cells per cross section was 6.7 ± 4.3 (1.4% of the total cell count) in the control group, 5.3 ± 3.7 (1.7%) in the PEG-DA treatment group, and 6.4 ± 4.7 (1.6%) in the PEG-L-DA treatment group. The number of inflammatory cells per cross section was 0.86 ± 0.90 (0.2% of the total cell count) in the control group, 0.71 ± 1.11 (0.2%) in the PEG-DA treatment group, and 0.43 ± 0.53 (0.1%) in the PEG-L-DA treatment group. In no case were inflammatory cells observed to be localized to the region of the hydrogel barrier.
Several control experiments were performed to investigate any direct influence upon arterial morphology that might have been induced by application of a hydrogel barrier. As a control on the possibility that the hydrogel barrier imposed nutritional limitations on the artery wall, the number of cells in the media was measured in noninjured vessels treated with a barrier for 28 days; medial cell number did not differ from that in control noninjured arteries (Table 2, P > 0.5). As a control on the possibility that the interfacial photopolymerization process damaged medial SMCs, medial cells were counted in noninjured vessels treated with a barrier for 2 days; medial cell number did not differ from that in control noninjured arteries (Table 2, P > 0.5). As a control on the possibility that the photopolymerization process altered intimal thickening, the hydrogel was formed around the periphery of arteries after balloon injury. The extent of intimal thickening at the 14th day and the medial cell number were not statistically different from those in arteries that were injured but not treated with a hydrogel in any way (intimal-to-medial area ratio of 1.06 ± 0.12 in animals with perivascular gel versus 0.92 ± 0.15 in control animals, P > 0.5; and 472 ± 51 cells per cross-section versus 460 ± 43, P > 0.5; comparing control in Table 3 to 14-day controls in Tables 1 and 2). That the hydrogel did not induce a foreign body response is established by the lack of inflammatory cell localization to the barrier, as described above.
Table 3.
Effects of local delivery of PDGF and thrombin upon intimal thickening and medial repopulation
| Group (n) | I/M* | Significance relative to control† | Number of medial cells/cross section | Significance relative to control‡ |
|---|---|---|---|---|
| Control, 14 days (7)§ | 1.06 ± 0.12 | 472 ± 51 | ||
| PEG-DA, 14 days (7) | 0.05 ± 0.02 | P < 0.001 | 306 ± 22 | P < 0.05 |
| PDGF + PEG-DA, 14 days (7) | 0.02 ± 0.01 | P < 0.001 | 297 ± 40 | P < 0.05 |
| PDGF, 14 days (7) | 2.27 ± 0.14 | P < 0.001 | 495 ± 25 | P > 0.4 |
| Thrombin + PEG-DA, 14 days (7) | 0.10 ± 0.08 | P < 0.001 | 282 ± 39 | P < 0.05 |
Values are mean ± SEM.
Ratio of the cross sectional areas of the intima and the media.
By the Mann–Whitney U test.
Control was injured, not treated with hydrogel barrier, but treated with a perivascular degradable hydrogel containing no protein.
Local Release of PDGF-BB and Thrombin.
The potential actions of PDGF-BB and thrombin were investigated by blocking postinjury thrombosis with a nondegradable hydrogel barrier and by adding back individually PDGF-BB and thrombin via local release from a perivascular degradable hydrogel (Table 3; Fig. 1 E and F). When the injured artery was exposed to blood, local release of PDGF-BB increased the extent of intimal thickening approximately 2-fold (P < 0.001), demonstrating that the protein was released in active form and at potent level. In the prolonged absence of blood contact obtained by treatment with a nondegradable barrier, the local release of PDGF-BB did not significantly increase intimal thickening (P > 0.5) or medial cell number (P > 0.5). Similarly, local release of thrombin did not stimulate intimal thickening (P > 0.5) or medial repopulation (P > 0.5).
bFGF Depletion.
Balloon injury has been shown to result in liberation of bFGF from medial stores, and this effect has been found to be maximal at 4 days (17). bFGF levels remaining in the vessel wall were assessed by ELISA 4 days postinjury in balloon-injured arteries with and without blood contact, as well as in noninjured arteries. Injury without blockade resulted in loss of 84% of medial bFGF compared with noninjured control arteries (0.23 ± 0.01 versus 0.04 ± 0.01, mean OD450 ± SEM normalized to total protein, P < 0.01), whereas blockade of blood contact prevented any statistically significant liberation of bFGF (0.19 ± 0.02, P > 0.5).
DISCUSSION
The objective of this study was to clarify the roles of selected luminal and medial factors in initiating the cascade of events leading to intimal thickening. It has been difficult in previous experiments to unequivocally separate the roles of luminal versus medial factors in intimal thickening, largely due to the experimental challenge of completely blocking the deposition of blood cells after injury. In the current study, we used a novel in situ photopolymerization technique to synthesize a thin, nonthrombogenic hydrogel barrier on the luminal surface of an artery. The nondegradable intravascular hydrogel barrier was found to remain on the arterial wall for the full duration of these experiments (28 days) with no change in thickness (P > 0.5). We also used a degradable hydrogel barrier, which blocked blood contact with the vessel wall for ≈24 hr. Thus, our experimental model permitted separation of medial and luminal components on two time scales: that of thrombosis (with the degradable barrier) and that of SMC migration, proliferation, and matrix secretion (with the nondegradable barrier). All experiments were performed after removal of the adventitia, permitting clear interpretation of the results in terms of medial and luminal activities as well as direct delivery of bioactive agents to the media from a perivascular depot. It should be understood that the injury model used in the rat in this study was more severe than might be observed in clinical balloon angioplasty, and that the artery in the clinical case is not removed of its adventitia. It is difficult to draw extrapolations between the clinical situation and this model in the rat for numerous reasons, not the least of which is the lack of an underlying vascular disease.
Several experiments were performed to probe the biocompatibility of the hydrogel barrier, namely whether it presented nutritional limitations to the artery wall, caused photochemical damage during hydrogel formation or induced an inflammatory reaction while present on the artery surface. While the hydrogel barrier blocked blood cell deposition following injury, it is relatively permeable to small-to-moderately sized proteins (19), so it only limited exposure to thrombus derived (versus circulating) proteins. The hydrogel barrier did not appear to interfere with the nutritional supply of the vessel wall, as the long term presence of the barrier did not alter the number of viable cells per cross-section even over durations as long as 28 days.
Control experiments were performed to probe the biocompatibility of the interfacial photopolymerization process of hydrogel formation that involves the action of free radicals. When the hydrogel barrier was applied in arteries without balloon injury, no medial cell death was noted at 2 days, even in the cellular layers in direct contact with the hydrogel, indicating that the photochemical process of barrier formation was not directly cytotoxic. This observation is in agreement with previous observations from our laboratory where we have used similar photochemical manipulations to encapsulate cell aggregates within polymeric hydrogels (18). When a hydrogel was formed perivascularly after balloon injury and removal of the adventitia, no statistically significant differences in medial cell number or intimal thickening were noted. Thus, as judged from these control experiments, the photochemical process of formation of the hydrogel barriers did not appear to alter arterial morphology in either the quiescent or healing state.
As a final measure of biocompatibility of the hydrogel barrier, cellular interactions with the material were investigated. The hydrogel barrier remained free of adherent platelets and other blood cells throughout the 28-day duration of blood contact; thus, no biological reaction was observed on the luminal side of the barrier. The localized presence of inflammatory cells was investigated through Giemsa staining as well as immunohistochemical identification of SMCs. No statistically significant differences in the number of inflammatory cells or non-SMCs within the arterial media were noted between the control group and those treated with PEG-DA barriers or PEG-L-DA barriers. Moreover, no preferential localization of the inflammatory cells or non-SMCs was noted at any site within the artery, including near the hydrogel barrier. Thus, the presence of the hydrogel barrier did not appear to induce any local or distributed foreign body reaction.
It is possible that a nondegradable hydrogel barrier might have acted exclusively as a mechanical barrier to prevent migration of SMCs across the internal elastic lamina regardless of the biochemical signals inducing SMC migration. Our hypothesis, however, is that the hydrogel barrier reduced intimal thickening by preventing thrombosis and thus exposure to mitogenic and chemotactic factors derived from the thrombus. To distinguish between these two possibilities, we performed the investigation with barriers that were present over two vastly different time scales, one on the scale of thrombosis (PEG-L-DA, present for ≈24 hr) and the other on the scale of SMC migration and proliferation (PEG-DA, present for the duration of the 14 or 28 day experiment). If the effect on intimal thickening was the result of blocking thrombus-derived bioactive factors, short-term blockade of blood contact should provide similar results with respect to intimal thickening as permanent blockade. There was no statistically significant difference in intimal thickening for groups treated with degradable (90% reduction at 14 days) versus nondegradable (95% reduction at 14 days, P > 0.2) hydrogel barriers. This observation supports the hypothesis that the hydrogel barrier acts by blocking early bioactive signals derived from thrombosis, but it does not support the alternative hypothesis that it acted by direct blockade of SMC migration across the internal elastic lamina. This observation also suggests that the hydrogel barriers do not impact intimal thickening by altering the transmission of hemodynamic shear stresses into the arterial wall, as remodeling to such changes would be expected to occur over a relatively long time period. Furthermore, the delayed medial repopulation, i.e., the reduced cell number of the media that was observed with PEG-DA barrier treatment, was not observed with PEG-L-DA barrier treatment. This suggests that the signals necessary for efficient medial repopulation are blood-derived, but not necessarily entirely thrombus-derived.
Balloon-induced arterial injury consists of two components: (i) removal of endothelial cells and (ii) stretch-induced damage to medial SMCs. Clowes et al. (20) used a model of deendothelialization without stretch injury or SMC death and demonstrated that endothelial disruption and platelet deposition alone were insufficient to promote intimal thickening. Our results demonstrate that stretch injury and SMC death is likewise insufficient to induce intimal thickening. In the absence of platelet deposition, intimal thickening was essentially absent, presumably due to a role played by a thrombus-associated factor such as PDGF or thrombin. To probe this possibility with these two proteins, we used a perivascular local release system to add back the PDGF or thrombin that would have been supplied by the thrombus had the hydrogel barrier been absent. If the intimal thickening response induced by normal postinjury exposure to blood was reconstituted by local release of PDGF-BB or thrombin, then a key causational role of that protein would be strongly supported.
Reconstitution of PDGF-BB or thrombin individually failed to induce intimal thickening or stimulate medial repopulation when postinjury blood contact was prohibited. In these experiments, the adventitia was removed from the arterial wall to facilitate diffusion of PDGF-BB and thrombin into the media (for consistency, the adventitia was removed in all cases). When PDGF-BB was delivered perivascularly after balloon injury in the normal presence of thrombosis and blood contact, intimal thickening was significantly exacerbated, confirming that PDGF-BB was delivered in bioactive form. While only one dose of PDGF-BB was evaluated [that being approximately twice the amount released by a platelet thrombus 50 μm thick (assuming that only approximately half of the protein in the perivascular hydrogel diffuses in the direction of the media)], the clear stimulation of intimal thickening in the positive control suggests that the PDGF dose examined is biologically relevant. The corresponding lack of a response to PDGF-BB when postinjury blood contact was prohibited suggests that PDGF-BB plays an important role in stimulating intimal thickening after the cascade of events has begun, but that it cannot initiate intimal thickening in the absence of some other luminally derived cofactor.
We may now conclude that PDGF, although necessary, is not sufficient to induce postinjury intimal thickening in the rat. Reidy et al. (6) used PDGF-neutralizing antibodies to demonstrate by depletion that PDGF plays a critical role in the intimal thickening process. Furthermore, Nabel et al. (21) demonstrated that expression of recombinant PDGF-B in uninjured porcine iliofemoral arteries resulted in an 8-fold increase in intimal thickening relative to control transfected arteries. It should be noted that in the study by Nabel et al. (21), blood contact was permitted, and other blood-derived factors may have been acting on the vessel wall, especially in view of the fact that a small degree of intimal thickening was observed in control transfected arteries. Our results demonstrate that elevated exposure to PDGF-BB exacerbates neointima formation after medial injury, but only when blood contact is permitted; some other luminally derived factor would appear to be required in conjunction with PDGF-BB. Similarly, locally released thrombin did not reconstitute the effects of platelet deposition. It was not possible to locally release thrombin in the absence of a luminal barrier, as this has been shown to elevate platelet deposition (22). We did not simultaneously reconstitute PDGF and thrombin, and thus our results do not address any possible synergy between these two factors; evidence suggests that PDGF and thrombin may act cooperatively (23, 24).
From the results of this study, we may now also conclude that the factors leading to depletion of bFGF from the media are not intrinsic to the arterial wall. Levels of both bFGF protein and mRNA have been shown to decrease in response to arterial injury and to remain low for prolonged time periods, suggesting that bFGF is an important signal for the induction of proliferation but not for the sustenance of proliferation (17). Blockade of blood contact with the hydrogel barrier prevented the depletion of bFGF from the vessel wall that is characteristically seen after balloon injury (17), suggesting that the bFGF has remained in an inactive form unable to act upon cell-surface receptors, and thus unable to impact arterial healing. Given that the hydrogel barrier is relatively permeable to moderately sized proteins (19), such as bFGF, the reduction in bFGF depletion would not appear to be due to mechanical entrapment of bFGF in the media by the barrier, but rather to the absence of a signal required for bFGF mobilization and liberation.
bFGF has been shown to be a potent stimulus for intimal thickening, sufficient to induce it even in the absence of intimal or medial injury (13, 25). Direct gene transfer of a plasmid encoding a secreted form of acidic FGF caused substantial intimal thickening in the absence of medial injury (26). bFGF does not contain a classic signal sequence and thus is not believed to be secreted (27), but rather leaked from damaged cells and sequestered in the extracellular matrix. We assayed the total amount of bFGF in the arterial wall and did not distinguish between intracellular and extracellular bFGF. Our results suggest that mobilization of bFGF from medial stores after vascular injury occurs only when blood contact is permitted, and thus that the actions of this important medial factor may be under the control of luminal factors. This luminal factor would appear to be thrombus-derived. Platelet heparitinase seems a plausible candidate; platelet heparitinase has recently been demonstrated to release active bFGF from endothelial cell-derived extracellular matrix in vitro (28, 29). Alternatively, thrombin has been shown to enhance degradation of heparan sulfate by tumor cell heparitinases in vitro (30), suggesting the possibility that a number of blood-derived factors may act together. Plasmin has also been shown to liberate active bFGF from the extracellular matrix in vitro (31), and exposure to plasmin during spontaneous thrombolysis may be involved in bFGF liberation in the injured artery. Future work will investigate the ability of blood-derived enzymes to liberate bFGF from the vessel wall after injury in the absence of blood contact and will determine if the liberation of bFGF is sufficient to efficiently repopulate the media and initiate the cascade of events leading to neointima formation.
Acknowledgments
The technical assistance of Kathleen Luther and Valerie Virta is greatly appreciated. We thank Focal, Inc. (Lexington, MA) for partial research funding.
Footnotes
Abbreviations: SMC, smooth muscle cell; bFGF, basic fibroblast growth factor; PDGF, platelet-derived growth factor; HBS, Hepes-buffered saline; PEG-DA, PEG diacrylate; PEG-L-DA, PEG-co-polylactic acid diacrylate.
References
- 1.Ip J H, Fuster V, Badimon L, Badimon J, Taubman M B, Chesebro J H. J Am Coll Cardiol. 1990;15:1667–1687. doi: 10.1016/0735-1097(90)92845-s. [DOI] [PubMed] [Google Scholar]
- 2.Hill-West J L, Chowdhury S M, Slepian M J, Hubbell J A. Proc Natl Acad Sci USA. 1994;91:5967–5971. doi: 10.1073/pnas.91.13.5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slepian M J, Massia S P, Weselcouch E, Khosravi F, Roth L. Circulation. 1995;92:3297. (abstr.). [Google Scholar]
- 4.Slepian M J, Massia S P, Weselcouch E, Khosravi F, Roth L. Circulation. 1995;92:1823. [Google Scholar]
- 5.Jawien A, Bowen-Pope D F, Lindner V, Schwartz S M, Clowes A W. J Clin Invest. 1992;89:507–511. doi: 10.1172/JCI115613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferns G A A, Raines E W, Sprugel K H, Motani A S, Reidy M A, Ross R. Science. 1991;253:1129–1132. doi: 10.1126/science.1653454. [DOI] [PubMed] [Google Scholar]
- 7.McNamara C A, Sarembock I J, Gimple L W, Fenton J W, Coughlin S R, Owens G K. J Clin Invest. 1993;91:94–98. doi: 10.1172/JCI116206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ip J H, Fuster V, Israel D, Badimon L, Badimon J, Chesebro J H. J Am Coll Cardiol. 1991;17:77–88. doi: 10.1016/0735-1097(91)90942-3. [DOI] [PubMed] [Google Scholar]
- 9.Sarembock I J, Gertz S D, Gimple L W, Owen R M, Powers E R, Roberts W C. Circulation. 1991;84:232–243. doi: 10.1161/01.cir.84.1.232. [DOI] [PubMed] [Google Scholar]
- 10.Schwartz R S, Holmes D R, Topol E J. J Am Coll Cardiol. 1992;20:1284–1293. doi: 10.1016/0735-1097(92)90389-5. [DOI] [PubMed] [Google Scholar]
- 11.Bashkin P, Doctrow S, Klagsbrun M, Svahn C M, Folkman J, Vlodavsky I. Biochemistry. 1989;28:1737–1743. doi: 10.1021/bi00430a047. [DOI] [PubMed] [Google Scholar]
- 12.Vlodavsky I, Bashkin P, Ishai-Michaeli R, Chajek-Shaul T, Bar-Shavit R, Haimovitz-Friedman A, Klagsbrun M, Fuks Z. Ann NY Acad Sci. 1991;638:207–222. doi: 10.1111/j.1749-6632.1991.tb49032.x. [DOI] [PubMed] [Google Scholar]
- 13.Edelman E R, Nugent M A, Smith L T, Karnovsky M J. J Clin Invest. 1992;89:465–473. doi: 10.1172/JCI115607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedman R J, Stemerman M B, Wenz B, Moore S, Gauldie J, Gent M, Tiell M L, Spaet T H. J Clin Invest. 1977;60:1191–1201. doi: 10.1172/JCI108872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ross R, Raines E W, Bowen-Pope D F. Cell. 1986;46:155–169. doi: 10.1016/0092-8674(86)90733-6. [DOI] [PubMed] [Google Scholar]
- 16.Wagner W R, Hubbell J A. J Lab Clin Med. 1990;116:636–650. [PubMed] [Google Scholar]
- 17.Olson N E, Chao S, Lindner V, Reidy M A. Am J Pathol. 1992;140:1017–1023. [PMC free article] [PubMed] [Google Scholar]
- 18.Pathak C P, Sawhney A S, Hubbell J A. J Am Chem Soc. 1992;114:8311–8312. [Google Scholar]
- 19.West J L, Hubbell J A. React Polym. 1995;25:139–147. [Google Scholar]
- 20.Clowes A W, Clowes M M, Fingerle J, Reidy M A. Lab Invest. 1989;60:360–364. [PubMed] [Google Scholar]
- 21.Nabel E G, Yang Z, Liptay S, San H, Gordon D, Haudenschild C C, Nabel G J. J Clin Invest. 1993;91:1822–1829. doi: 10.1172/JCI116394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blomback B, Meier J, Stocker K. Thromb Haemostasis. 1995;73:1469. [Google Scholar]
- 23.Soyombo A A, Dicorleto P E. J Biol Chem. 1994;269:17734–17740. [PubMed] [Google Scholar]
- 24.Shankar R, Delamotte C A, Poptic E J, Dicorleto P E. J Biol Chem. 1994;269:13936–13941. [PubMed] [Google Scholar]
- 25.Lindner V, Lappi D A, Baird A, Majack R A, Reidy M A. Circ Res. 1991;68:106–113. doi: 10.1161/01.res.68.1.106. [DOI] [PubMed] [Google Scholar]
- 26.Nabel E G, Yang Z, Plautz G, Forough R, Zhan X, Haudenschild C C, Maciag T, Nabel G J. Nature (London) 1993;362:844–846. doi: 10.1038/362844a0. [DOI] [PubMed] [Google Scholar]
- 27.Burgess W H, Maciag T A. Annu Rev Biochem. 1989;58:575–606. doi: 10.1146/annurev.bi.58.070189.003043. [DOI] [PubMed] [Google Scholar]
- 28.Presta M, Maier J A M, Rusnati M, Ragnoti G. J Cell Physiol. 1989;140:68–74. doi: 10.1002/jcp.1041400109. [DOI] [PubMed] [Google Scholar]
- 29.Ishai-Michaeli R, Eldor A, Vlodavsky I. Cell Regul. 1990;1:833–842. doi: 10.1091/mbc.1.11.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benezra M, Vlodavsky I, Bar-Shavit R. Exp Cell Res. 1992;201:208–215. doi: 10.1016/0014-4827(92)90365-f. [DOI] [PubMed] [Google Scholar]
- 31.Saksela O, Rifkin D B. J Cell Biol. 1990;110:767–775. doi: 10.1083/jcb.110.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]