

Abstract
The congenital long QT syndrome (LQTS) is an inherited disorder characterized by a prolonged cardiac action potential. This delay in cellular repolarization can lead to potentially fatal arrhythmias. One form of LQTS (LQT3) has been linked to the human cardiac voltage-gated sodium channel gene (SCN5A). Three distinct mutations have been identified in the sodium channel gene. The biophysical and functional characteristics of each of these mutant channels were determined by heterologous expression of a recombinant human heart sodium channel in a mammalian cell line. Each mutation caused a sustained, non-inactivating sodium current amounting to a few percent of the peak inward sodium current, observable during long (>50 msec) depolarizations. The voltage dependence and rate of inactivation were altered, and the rate of recovery from inactivation was changed compared with wild-type channels. These mutations in diverse regions of the ion channel protein, all produced a common defect in channel gating that can cause the long QT phenotype. The sustained inward current caused by these mutations will prolong the action potential. Furthermore, they may create conditions that promote arrhythmias due to prolonged depolarization and the altered recovery from inactivation. These results provide insights for successful intervention in the disease.
Keywords: SCN5A, LQT3, sodium channel, heart, hH1
The long QT syndrome (LQTS) is an inherited disorder characterized by a delay in cardiac cellular repolarization (1, 2). This cellular based delay in repolarization is manifest macroscopically as a prolongation of the QT interval of the surface electrocardiogram in patients with the disease. The molecular basis of the disease is heterogeneous (3, 4). One LQT locus (LQT3) on human chromosone 3 encodes the voltage-gated cardiac sodium channel α subunit (hH1 and SCN5A; refs. 5 and 6). Three distinct mutations in SCN5A were identified in this subset of LQT patients (4, 7). These include an in-frame deletion of three amino acids (Lys-1505, Pro-1506, and Glu-1507, ΔKPQ), and two point mutations, N1325S (Asp-1325 converted to serine) and R1644H (Arg-1644 converted to histidine). A partial characterization of the single channel properties of these mutant channels expressed in Xenopus laevis oocytes has recently been described (8, 9). The purpose of this study was to elucidate and compare the whole-cell properties of the three known sodium channel mutations that are linked to the LQTS by heterologous expression in a human cell line.
MATERIALS AND METHODS
Voltage Clamp Methods.
The methods used have been described previously (10–12). Briefly, macroscopic sodium currents were recorded using the whole-cell method of the patch clamp technique (13). Electrodes resistances ranged from 0.8 to 2 MΩ. Voltage clamp command pulses were generated using pclamp software (version 6.0, Axon Instruments, Foster, CA). Currents were filtered at 5 kHz (−3 dB, 4 pole Bessel filter). An Axopatch 200 patch clamp amplifier was used with series resistance compensation. The standard holding potential for all pulse protocols was −120 mV. Data are presented as mean ± SEM. Experiments were carried out at room temperature (20–22°C).
Solutions: Whole-Cell Recording.
The bath solution contained 145 mM NaCl, 4 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 10 mM Hepes, and 10 mM glucose (pH 7.35). The pipette solution contained (intracellular solution) 10 mM NaF, 110 mM CsF, 20 mM CsCl, 10 mM EGTA, and 10 mM Hepes (pH 7.35).
Site-Directed Mutagenesis.
Site-directed mutagenesis of hH1 was performed using either the pALTER system (Promega) or PCR mutagenesis. For construction of N1325S, a 1998-bp EcoRI/KpnI fragment of hH1 (nt 2231–4228) was subcloned into the pALTER-1 plasmid and antisense single-strand DNA rescued using the R408 helper phage. Mutagenesis was carried out using a phosphorylated sense oligonucleotide (5′-atgagggtggtggtcagcgcgctggtgggcgccatc-3′) according to the plasmid supplier’s protocol. Mutant recombinants were identified by digestion with BssHII (site created by the N1325S mutation in combination with a silent nucleotide change in codon 1326). Multiple independent recombinants were subcloned, resequenced to verify the mutation, and used for expression studies. The KPQ mutation has been described (8).
For construction of R1644H, a single step PCR mutagenesis procedure was employed. A forward primer (5′-gccaaggggatccacacgctgctctttgccctcat-3′) which incorporates the R1644H mutation and overlaps a natural BamHI site at nt 4926 was paired with a reverse primer (5′-ccattgctgttgggcagagt-3′) located 3′ to an EagI site at nt 5130. Reactions (100 μl) contained the following: 100 ng pSP64T-hH1 plasmid DNA, 0.5 μM each primer, 0.2 mM each deoxynucleoside triphosphate, 10 mM Tris·HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, and 2.5 units of Taq polymerase (Boehringer Mannheim). Amplifications were carried out for 20 cycles of 94°C for 1 min, 54°C for 1 min, and 72°C for 1 min. Products (409 bp) were purified by spin column chromatography (Qiagen, Chatsworth, CA), digested with BamHI and EagI, and subcloned into full-length pSP64T-hH1 and tested for expression in oocytes. Functional clones were then subcloned into pRc/CMV for expression in mammalian cells. Multiple independent recombinants were isolated and sequenced thoroughly in the region amplified by PCR to identify polymerase errors. Three independent clones were tested for each mutant to confirm the phenotype.
Transfection of hH1 and Mutant hH1 cDNA for Expression in tsA-201 Cells.
Transient transfection with calcium phosphate was used to express mutant and wild-type (WT) hH1 in a transformed human kidney cell line (HEK 293; tsA-201) stably expressing the simian virus 40 T-antigen. Cell transfection was carried out using 10 μg of plasmid DNA encoding channel and 2 μg of plasmid encoding green fluorescent protein (GFP). GFP cotransfection was used to visually identify cells expressing exogenous channel DNA (12).
RESULTS
Fig. 1 compares each of the LQT3 causing Na+ channel mutations to the WT channel. In these experiments voltage clamp pulses were applied to −20 mV to activate Na+ channels in WT or in cells expressing the three different LQT3 Na+ channel mutations. Plotted above each current recording is the ensemble variance obtained by averaging multiple current records. The variance is generated by channel gating fluctuations (14) and provides a sensitive measure of channel gating activity. The ensemble current and variance both declined to zero within 10 msec in the WT channels. In contrast, the LQT3 mutations caused the channels to continue opening and closing throughout the depolarization step. The ΔKPQ, R1644H, and N1325S mutants all showed sustained inward current and non-zero variance, indicating continued channel gating, at times later than 20 msec. Interestingly, there were no gross changes in channel kinetics. We examined this in more detail by measuring several kinetic parameters including the time to peak sodium current, the time course of inactivation, and the time course of recovery from inactivation.
Figure 1.
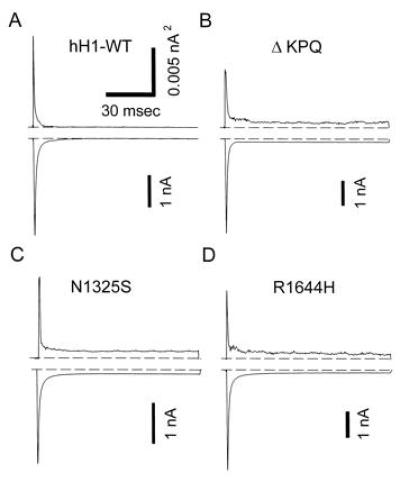
Whole-cell recording of WT, ΔKPQ, N1325S, and R1644H human heart Na+ channels expressed in HEK293/tsA201 cells. Voltage clamp steps of 100 ms duration to −20 mV from a holding potential of −120 mV were applied every 5 sec. The average of 20 consecutive recordings is shown along with the variance computed during the averaging process. The nonstationary variance is a sensitive indicator of gating. Note the non-zero variance that occurs after 20 msec in the mutant channels. The ensemble variance, σ2, is related to channel gating according to the following relationship: σ2 = P (1 − P) N i2, where P is the open probability, N is the number of channels, and i is the single channel current. N and i are constants at a particular membrane potential. The variance is a sensitive measure of continued channel gating. As channels inactivate, the variance and current drop to zero. The mutant channels show a sustained sodium current that persists after the WT channels have inactivated. This late gating activity is reflected in the non-zero variance that exists during the prolonged depolarizations.
The time-to-peak sodium current was measured for WT (777 ± 35 μsec, n = 23) and each mutant at −20 mV. A significant decrease (P < 0.01) was observed only for the R1644H mutant (638 ± 22 μsec, n = 22) suggesting that either the rate of activation or inactivation was enhanced in this mutant. There was no significant change (P > 0.1) compared with WT in time-to-peak sodium current for ΔKPQ (709 ± 22 μsec, n = 22) or N1325S (766 ± 33 μsec, n = 23).
Inactivation Kinetics.
The time course of channel inactivation was estimated quantitatively by fitting the declining phase of inward sodium current with both single and double exponential functions. The double exponential function gave the best fit to the data both by visual and statistical estimation. In WT hH1 the two time constants of macroscopic inactivation at −20 mV were 0.6 ± 0.04 and 4.9 ± 1.25 msec (n = 5), respectively. Approximately 80–90% of the Na+ current inactivates with the smaller time constant (faster process). Fig. 2 summarizes the voltage dependence of these time constants and the relative amplitudes of each component for each mutant channel. The relative amplitude (in percent) for each of the exponential components is shown in Fig 2 A–C. In all cases there was a large amplitude component that decayed with the smaller time constant (τfast = 0.6–2 msec). The steady-state component, labeled Alate, is plotted as triangles. These show the average amount and voltage dependence of this sustained component for each mutant channel. Fig. 2 D–F show the averaged time constants for these fits. The average time constants for the wild type channels are shown as dashed lines (11). Statistically significant differences between WT and a mutant are denoted by asterisks. The fraction of the sustained late Na+ current was dependent on membrane potential and was different for each mutant. In the ΔKPQ mutant channel, the fraction of sustained current decreased from about 5% at −40 mV to ≈2% at +40 mV. At a membrane potential of −20 mV, the sustained inward sodium current was 2.57 ± 0.27% (n = 13) of the peak sodium current. In contrast, the N1325S mutant showed the opposite voltage dependence for the late openings, increasing from 2.5% at −40 mV to 5% at +40 mV. The mean sustained inward current at −20 mV was 3.12 ± 0.65% (n = 8). Finally the R1644H mutant Na+ channel showed almost no voltage dependence with a constant sustained fraction of inward current of approximately 0.8% between −40 and +30 mV and an average at −20 mV of 0.80 ± 0.14% (n = 8).
Figure 2.
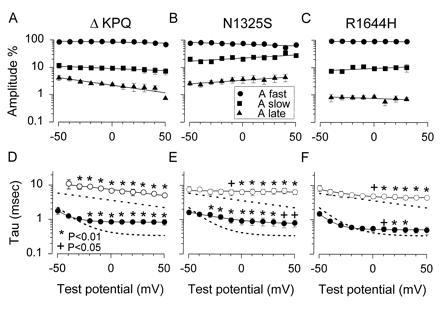
Amplitudes and time constants of decay of Na+ current in control and in LQT mutant channels. (A–C) Relative amplitudes of the inactivating sodium current. The fraction of non-inactivating current is plotted as triangles and labeled Alate. The time course of inactivation of mutant hH1 channels was resolved into two components by fitting the decreasing phase of the inward sodium current (inactivation) with an equation containing the sum of two exponentials plus a baseline. (D–F) Fitted time constants. The dashed lines indicate the voltage dependence of WT hH1 time constants (11). Significant differences are indicated by asterisks or plus signs (∗, +). Symbols represent means, with error bars indicating the standard error of the mean.
The fractional amplitude of the fast component was greater in the ΔKPQ and R1644H mutants than in WT (not shown). The second, slower component was consequently smaller. This gave the visual impression of a more rapidly decaying sodium current compared with control. Indeed when a single exponential function was fitted to the data (not shown), the time constants were smaller (apparent faster decay). However, a two exponential fit was statistically better (F-ratio test; P < 0.05) in all cases. The time constants at several potentials (see Fig. 2) were statistically significantly larger than WT.
Recovery from Inactivation.
Since each mutation appeared to alter channel inactivation, we investigated recovery from inactivation using a two-pulse protocol. The first pulse which placed all channels in the inactivated state was followed by a second voltage clamp step to −20 mV to assess the fraction of channels that had recovered from inactivation following various time intervals at −120 mV. These intervals are plotted on the abscissa in Fig. 3. The recovery curves were fitted with an exponential function containing two or three components to quantify the recovery relationship. The curves were fit to the data averaged from multiple cells (see Figure legend). The best fitting exponential functions are shown as solid curves in Fig. 3. In the WT channels the largest fraction (77%) recovered with a time constant of 6.7 msec. A second smaller component (23%) recovered with a time constant of 35 msec.
Figure 3.
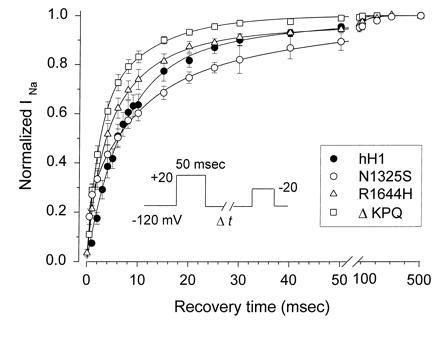
Recovery from inactivation in WT and LQT mutant Na+ channels. Channels were inactivated by a 50-msec prepulse to +20 mV. The fraction of channels that had recovered following various time intervals at −120 mV (shown on the abscissa) was calculated by dividing the peak current measured during a test pulse to −20 mV by the current measured (at −20 mV) after clamping the cell for 10 sec at −120 mV. The number of cells use to obtain each mean was 12 (WT hH1), 11 (ΔKPQ), 7 (R1644H), and 6 (N1325S). The averaged data were fitted with 1-, 2-, and 3-exponential functions. The best fitting curves for WT and each mutant are shown as solid lines. WT and ΔKPQ were best fitted with a 2-exponential function and there was no significant improvement in the fit when a third component was included. R1644H and N1325S were both best fitted by a 3-exponential function. The best fitting parameters from these two exponential fits were as follows (τ1, A1, τ2, A2, τ3, A3): WT, 6.7 msec, 77%, 35 msec, 23%; ΔKPQ, 2.1 msec, 59%, 11 msec, 41%; N1325S, 1.3 msec, 40%, 25 msec, 60%; R1644H, 3.3 msec, 68%, 26 msec, 32%. The N1325S and R1644H mutant recoveries were fitted significantly better by a 3 exponential function (P < 0.05; F-ratio test). The fitted parameters were as follows: N1325S, 0.5 msec, 24%, 8.2 msec, 43%, 44 msec, 33%; R1644H, 2 msec, 36%, 8.3 msec, 52%, 66 msec, 12%. Since the functions were fitted to the averaged data, error estimates on these parameters were not obtained, but the experimental errors are plotted with the data.
The ΔKPQ mutant showed significantly faster overall recovery than WT channels. The R1644H mutant recovered from inactivation at −120 mV approximately the same as WT channels. Both R1644H and N1325S were significantly better fitted by a 3-exponential function. Recovery in the N1325S mutant channel was significantly slower than the WT and the other mutants. One component recovered very rapidly, but a third component caused N1325S to recover slower than WT. Although two of the mutants showed recoveries that differed from WT channels, it should be noted that the channels were all fully recovered from inactivation within ≈100–200 msec (see Fig. 3).
Steady-State Activation, Inactivation, and “Window Currents.”
A possible mechanism for a sustained late current is an overlap in the relationships for channel activation and inactivation. Such an overlap has been referred to as a window current. A sustained current will be observed if there is a fraction of channels that open (Popen > 0) when the probability for inactivation is also not zero. We evaluated this possibility by constructing steady-state inactivation curves and compared them to the curves for the voltage dependence of relative Na+ conductance. The voltage dependence of relative Na+ conductance was measured by using a series of voltage clamp steps to different membrane potentials between −90 and +40 mV. The holding potential was −120 mV. The peak current measured at each membrane potential was divided by the driving force for sodium ions (V − ENa) and the maximum sodium conductance. The steady-state inactivation relationships were obtained by measuring the peak sodium current during a step to −20 mV which followed a 500-msec prepulse to membrane potentials between −160 and −50 mV. The current during the test step was normalized by the maximum current measured following the prepulse to −160 mV. Each curve was fitted with a Boltzmann equation to estimate the membrane potential for half maximal activation and inactivation (V½), and to estimate the slope factors (k) for these relationships. The normalized conductance-voltage and steady-state inactivation relationships are plotted in the Insets in Fig. 4 A–D. The WT curves are also shown in each panel as dashed lines. The main graphs show the regions of overlap on an expanded scale. The fitted parameters are provided in Table 1.
Figure 4.
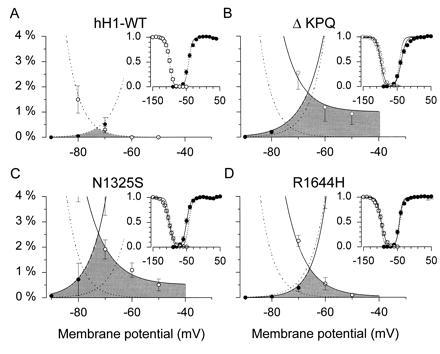
Steady-state inactivation and window current. Comparison of steady-state (500-msec) inactivation and activation curves for hH1 and LQT mutant sodium channels. A two-pulse protocol was used to estimate the membrane potential dependence of inactivation. A 500-msec prepulse was applied to different membrane potentials as shown on the abscissa. The fraction of channels not inactivated by the prepulse was estimated from the peak inward current during a test step to −20 mV. The peak inward current at each prepulse potential was normalized by the peak sodium current obtained following a prepulse to −160 mV where all channels are available to open. The channel activation curve was estimated by measuring peak sodium current during different test steps from a holding potential of −120 mV. The current at each membrane potential was divided by the electrochemical driving force for sodium ions, Vm − VNa. VNa represents the sodium equilibrium potential. The curves shown was normalized by the maximum sodium conductance. (Insets) Data over the entire range of membrane potentials tested. The main graphs show an enlargement of the window region and the data are converted to percentages. The shaded area represents the window region. Data are shown as means and standard errors (n = 7–24 cells in each curve).
Table 1.
Parameters of inactivation and activation
| WT, mV | ΔKPQ, mV | R1644H, mV | N1325S, mV | |
|---|---|---|---|---|
| Inactivation | ||||
| V½ | −99.6 ± 0.88 | −96.0 ± 1.9 | −97.5 ± 2.2 | −102.1 ± 2.0 |
| Slope factor, k | 5.72 ± 0.33 | 6.4 ± 0.3 | 6.9 ± 0.3* | 6.5 ± 0.4 |
| n | 11 | 11 | 7 | 13 |
| Activation | ||||
| V½ | −43.2 ± 1.9 | −37.2 ± 1.6* | −41.5 ± 2.2 | −49.6 ± 1.6* |
| Slope factor, k | 6.7 ± 0.33 | 8.6 ± 0.47* | 5.6 ± 0.44 | 5.7 ± 0.4 |
| n | 13 | 24 | 21 | 20 |
Statistically significant difference compared to WT, P < 0.05.
No significant differences were noted in the relationship between membrane potential and the steady-state availability for opening of WT and LQT mutant Na+ channels using 500-msec prepulses. The slope factor of the R1644H mutant was significantly (P < 0.05) changed compared with the WT. The relationships for the DKPQ and N1325S mutants differed significantly from WT (P < 0.05) but were shifted in opposite directions. The DKPQ mutant was shifted in the depolarizing direction by 6 mV. The slope factor of this relationship was also significantly changed from control. The curve for the N1325S mutant was shifted in the hyperpolarizing direction by 6.4 mV, but slope factor was not changed from WT.
These data demonstrate that there is indeed an overlap in these relationships that is greater in the mutants than in the WT channel. The overlap was maximal near −70 mV; however, for two of the mutants (N1325s and ΔKPQ), the steady-state inactivation relationship did not go to zero at the most depolarized potentials tested (−50 mV). The overlap in these relationships can in part account for the LQT phenotype; however, the late openings occur at more positive membrane potentials outside the range of the window in all three mutants. It is possible that this small current at −70 mV would depolarize cells with resting potentials in this range and lead to enhanced excitability.
Voltage Ramps.
To further characterize the steady state late sodium current, the membrane potential was ramped over 2 sec from −120 mV to +40 mV (Fig. 5). These slow membrane depolarizations largely inactivated WT channels before they could open (Fig. 5A). However, as seen in Fig. 5 B–D, inward sodium current was generated during these ramps for each of the mutant channels. These voltage ramp protocols directly demonstrate the non-inactivating nature of the mutant sodium channels. This non-inactivating inward current can produce significant depolarization late in the cardiac action potential plateau. This in turn will prolong the action potential and may promote conditions for subsequent inappropriate depolarizations and arrhythmias.
Figure 5.
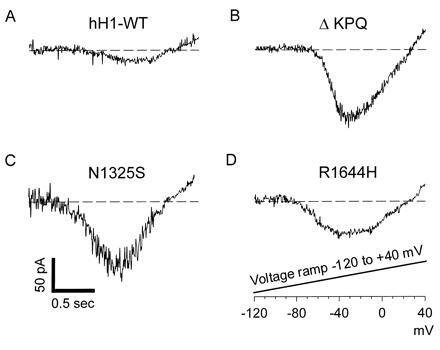
Voltage ramps to assess the non-inactivating sodium current of LQT mutant sodium channels. Cells were voltage clamped to a holding potential of −120 mV to assure all sodium channels were available to open. The membrane potential was slowly ramped from −120 mV to +40 mV over the course of 2 sec. This slow depolarizing ramp causes inactivation of WT channels, but non-inactivating LQT mutant channels continue to open and reveal the current voltage relationship of these channels.
DISCUSSION
This study demonstrates the macroscopic phenotype and properties of the three known LQT3 mutant sodium channels: ΔKPQ, N1325S, and R1644H. The results confirm and extend the observations on the ΔKPQ mutant channel obtained following heterologous expression in Xenopus oocytes (8, 9). They further demonstrate the properties of these mutants in a mammalian (human) cell line. A characterization of channel behavior in mammalian cells is important because previous studies have found anomalous behavior of some channels expressed in oocytes.
Possible mechanisms for the LQT phenotype include a Na+ current that persists throughout a depolarization. This could occur because of slowing of the rate of inactivation or because the inactivation process no longer goes to completion (15, 16). These data demonstrate that a change in the rate of decay of the macroscopic current does not fully account for the LQT defect. We have previously shown that the ΔKPQ mutation appeared to cause late opening behavior by randomly entering an altered gating mode where the transitions back into the open state from the inactivated state were temporarily increased (8). This transient loss of inactivation occurred ≈4% of the time and was sufficient to cause a late sustained sodium current that causes action potential prolongation and ultimately the long QT syndrome
Molecular Interpretation.
A simple cartoon of these LQT3 defects invokes an altered ability of the channel inactivation gate to remain closed (see refs. 17–19 for reviews). Previous studies have implicated the III–IV interdomain of the Na+ channel in inactivation (20–22). A deletion in this region, as occurs in the ΔKPQ mutant, could potentially alter the conformation of this putative gate and perturb its ability to successfully dock with its binding site. The N1325S mutation occurs in the S4–S5 region of domain III. This region is absolutely conserved between the heart, skeletal muscle, and rat brain IIa isoforms suggesting its importance (5, 23–25). The S4–S5 domain of voltage-gated potassium channels has been implicated as a binding site for the inactivation peptide in that channel (26). It is reasonable to speculate that this region performs a similar function in voltage-gated sodium channels. Presumably, the point mutation in this region destablizes binding of the inactivation gate.
The R1644H mutation is predicted to be near the cytoplasmic end of the S4 segment of domain IV. The positively charged S4 segment has been implicated in channel gating as a putative voltage sensor (or activation gate). Interestingly, this mutation had a significantly earlier time to peak sodium current compared with WT. This could occur either because channel inactivation or channel activation occurred more rapidly. As seen in Fig. 2, the time course of inactivation was not faster, suggesting an increase in the rate of activation in this mutant. In addition this mutant showed the smallest disruption of inactivation (see Figs. 2, 4, and 5). As noted above the S4–S5 segment may act as a binding site for the inactivation gate. Thus, the proximity of R1644H to this region may explain its effect on inactivation. The mutation may alter the structure of this region and interfere with successful binding of the inactivation gate to its receptor site. In addition, other studies have noted changes in channel inactivation by mutations in the S4 domain (27), suggesting that channel gating properties are not easily localized to specific domains. Of course, our data do not prove this simplistic model, and other less mechanical interpretations are possible and perhaps even likely. For example, these mutations may exert their effects allosterically with each mutation causing a convergent disruption of an inactivation mechanism that we do not yet fully understand.
Comparison with Skeletal Muscle Diseases.
Functional disturbances in another voltage-gated sodium channel are responsible for hereditary disorders of skeletal muscle membrane excitability. In hyperkalemic periodic paralysis, and certain other nondystrophic myotonias, missense mutations in the gene encoding the human skeletal muscle sodium channel α subunit (SCN4A, 17q), also cause disturbances in channel inactivation (28–31). Sodium channel mutations causing hyperkalemic periodic paralysis exhibit a biophysical phenotype that is similar to that exhibited by the LQT mutations studied here. Skeletal muscle sodium channel mutations in either the DII/S6 or DIV/S6 segments cause a small, persistent current to occur late in test depolarizations. In skeletal muscle, this sodium channel disturbance is believed to cause episodic, slow depolarization of the sarcolemma, leading eventually to conduction failure in the muscle fiber (28, 32). This disturbance also renders the muscle membrane hyperexcitable during periods in which the membrane depolarization is less severe. Additional mutations in the skeletal muscle sodium channel that cause the disease paramyotonia congenita exhibit more severe defects in inactivation (28, 29). Specifically, these mutations cause slowing of the macroscopic inactivation rate with a concomitant acceleration of recovery from inactivation. In skeletal muscle, these functional disturbances lead to sarcolemmal hyperexcitability and the clinical phenotype of myotonia. Such severe inactivation defects in the cardiac sodium channel would likely be incompatible with life.
Therapeutic Implications.
These experiments extend the biophysical evidence that the ΔKPQ, R1644H, and N1325S sodium channel mutations cause the long QT syndrome. Previously, it was demonstrated that these mutations lead to a small partial disruption of inactivation in channels expressed in Xenopus oocytes (8, 9). The data obtained from channels expressed in a human cell line demonstrate the generality of the original observations and reveal novel properties of each of the mutant channels. Each mutation produces a sustained inward sodium current that is predicted to account for the long QT defect. Computer-simulated action potentials (not shown) as well as studies with toxins that delay sodium channel inactivation indicate that these small increases in inward current are sufficient to prolong the cardiac action potential. Nevertheless, final proof must await direct measurements in transfected cardiac myocytes.
Additional novel findings are the distinct voltage dependencies of the late current in the different mutants, the window currents that exist, and the altered recoveries from inactivation. It is uncertain whether the altered recoveries would markedly affect cellular excitability or conduction times. In principle, these properties could precipitate the arrhythmias that accompany this disease (33). Finally the fraction of sustained late current was greatest in the N1325S mutant (≈3%), it was slightly less in the ΔKPQ mutant (≈2.6%) and was the smallest in the R1644H mutant (0.8%; all values at −20 mV). This suggests that the R1644H variant of the LQTS may be less severe than the other forms.
Acknowledgments
This work was supported by Grants HL51197, HL46681, and NS32387 from the National Institutes of Health. A.L.G. is a Lucille P. Markey Scholar. P.B.B. is an Established Investigator of the American Heart Association.
Footnotes
Abbreviations: LQT, long QT; LQTS, LQT syndrome; WT, wild type; ΔKPQ, Lys-1505, Pro-1506, and Glu-1507; N1325S, Asp-1325 converted to serine; R1644H, Arg-1644 converted to histidine.
References
- 1.Schwartz P J, Periti M, Malliani A. Am Heart J. 1975;89:378–390. doi: 10.1016/0002-8703(75)90089-7. [DOI] [PubMed] [Google Scholar]
- 2.Towbin J A, Li H, Taggart R T, Lehmann M H, Schwartz P J, Satler C A, Ayyagari R, Robinson J L, Moss A J, Hejtmancik J F. Circulation. 1994;90:2635–2644. doi: 10.1161/01.cir.90.6.2635. [DOI] [PubMed] [Google Scholar]
- 3.Curran M E, Splawski I, Timothy K W, Green E D, Vincent G M, Keating M T. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 4.Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson J L, Moss A J, Towbin J A, Keating M T. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 5.Gellens M E, George A L, Chen L, Chahine M, Horn R, Barchi R L, Kallen R G. Proc Natl Acad Sci USA. 1992;89:554–558. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.George A L, Varkony T A, Drabkin H A, Han J, Knops J F, Finley W H, Brown G B, Ward D C, Haas M. Cytogenet Cell Genet. 1995;68:67–70. doi: 10.1159/000133892. [DOI] [PubMed] [Google Scholar]
- 7.Wang Q, Shen J, Li Z, Timothy K, Vincent G M, Priori S G, Schwartz P J, Keating M T. Hum Mol Genet. 1995;4:1603–1607. doi: 10.1093/hmg/4.9.1603. [DOI] [PubMed] [Google Scholar]
- 8.Bennett P B, Yazawa K, Makita N, George A L. Nature (London) 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 9.Dumaine R, Wang Q, Keating M T, Hartmann H, Schwartz P J, Bown A M, Kirsch G E. Circ Res. 1996;78:916–924. doi: 10.1161/01.res.78.5.916. [DOI] [PubMed] [Google Scholar]
- 10.Valenzuela C, Bennett P B. Biophys J. 1994;67:161–171. doi: 10.1016/S0006-3495(94)80465-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang D W, George A L, Bennett P B. Biophys J. 1996;70:238–245. doi: 10.1016/S0006-3495(96)79566-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang D W, Nie L, George A L, Bennett P B. Biophys J. 1996;70:1700–1708. doi: 10.1016/S0006-3495(96)79732-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamill O P, Marty A, Neher E, Sakmann B, Sigworth F J. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 14.Sigworth F J. J Physiol (London) 1980;307:97–129. doi: 10.1113/jphysiol.1980.sp013426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aldrich R W, Corey D P, Stevens C F. Nature (London) 1983;306:436–441. doi: 10.1038/306436a0. [DOI] [PubMed] [Google Scholar]
- 16.Yue D T, Lawrence J H, Marban E. Science. 1989;244:349–352. doi: 10.1126/science.2540529. [DOI] [PubMed] [Google Scholar]
- 17.Patlak J. Physiol Rev. 1991;71:1047–1076. doi: 10.1152/physrev.1991.71.4.1047. [DOI] [PubMed] [Google Scholar]
- 18.Hille B. Ionic Channels of Excitable Membranes. 2nd Ed. Sunderland, MA: Sinauer; 1992. [Google Scholar]
- 19.Catterall W A. Annu Rev Biochem. 1995;64:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- 20.Stuhmer W, Conti F, Suzuki H, Wang X D, Noda M, Yahagi N, Kubo H, Numa S. Nature (London) 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- 21.Patton D E, West J W, Catterall W A, Goldin A L. Proc Natl Acad Sci USA. 1992;89:10905–10909. doi: 10.1073/pnas.89.22.10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.West J W, Patton D E, Scheuer T, Wand Y, Goldin A L, Catterall W A. Proc Natl Acad Sci USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noda M, Ikeda T, Kayano T, Suzuki H, Takeshima H, Kurasaki M, Takahashi H, Kuno M, Numa S. Nature (London) 1986;322:826–828. doi: 10.1038/322826a0. [DOI] [PubMed] [Google Scholar]
- 24.George A L, Komisarof J, Kallen R G, Barchi R L. Ann Neurol. 1992;31:131–137. doi: 10.1002/ana.410310203. [DOI] [PubMed] [Google Scholar]
- 25.Trimmer J S, Cooperman S S, Tomiko S A, Zhou J Y, Crean S M, Boyle M B, Kallen R G, Sheng Z H, Barchi R L, Sigworth F J, Goodman R H, Agnew W S, Mandel G. Neuron. 1989;3:33–49. doi: 10.1016/0896-6273(89)90113-x. [DOI] [PubMed] [Google Scholar]
- 26.Isacoff E Y, Jan Y N, Jan L Y. Nature (London) 1991;353:86–90. doi: 10.1038/353086a0. [DOI] [PubMed] [Google Scholar]
- 27.Yang N, George A L, Jr, Horn R. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]
- 28.Yang N, Ji S, Zhou M, Ptacek L J, Barchi R L, Horn R, George A L., Jr Proc Natl Acad Sci USA. 1994;91:12785–12789. doi: 10.1073/pnas.91.26.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chahine M, George A L, Jr, Zhou M, Ji S, Sun W, Barchi R L, Horn R. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 30.Cummins T R, Zhou J, Sigworth F J, Ukomadu C, Stephan M, Ptacek L J, Agnew W S. Neuron. 1993;10:667–678. doi: 10.1016/0896-6273(93)90168-q. [DOI] [PubMed] [Google Scholar]
- 31.Cannon S C, Strittmatter S M. Neuron. 1993;10:317–326. doi: 10.1016/0896-6273(93)90321-h. [DOI] [PubMed] [Google Scholar]
- 32.Cannon S C, Brown R H, Jr, Corey D P. Biophys J. 1993;65:270–288. doi: 10.1016/S0006-3495(93)81045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwartz P J, Priori S G, Locati E H, Napolitano C, Cantu F, Towbin J A, Keating M T, Hammoude H, Brown A M, Chen L K, Colatsky T J. Circulation. 1995;92:3381–3386. doi: 10.1161/01.cir.92.12.3381. [DOI] [PubMed] [Google Scholar]