

Abstract
To study the evolution of the yeast protein interaction network, we first classified yeast proteins by their evolutionary histories into isotemporal categories, then analyzed the interaction tendencies within and between the categories, and finally reconstructed the main growth path. We found that two proteins tend to interact with each other if they are in the same or similar categories, but tended to avoid each other otherwise, and that network evolution mirrors the universal tree of life. These observations suggest synergistic selection during network evolution and provide insights into the hierarchical modularity of cellular networks.
Biological networks are the basis of cellular functions (1, 2). Understanding network evolution may shed light on the hierarchical modularity, scale-free property, and various uses of the building blocks of biological networks (3–12). The yeast protein interaction network is one of the best annotated complex networks to date (13–17). Previous studies on the evolution of this network focused either on gene duplication and molecular evolution at the protein level (9, 10) or on the global statistical properties (12). Neither approach can delineate the network evolutionary path, and there is no other comparable protein interaction data for the system-level comparison approach (5). Therefore, uncovering the growth patterns and the evolutionary path of the protein interaction network is a serious challenge (3, 4, 6, 7, 9, 12).
Parts of the present yeast protein interaction network would have been inherited from the last common ancestor of the three domains of life: Eubacteria, Archaea, and Eukaryotes. Thus, an analysis of the evolution of the yeast protein interaction network may provide new insights into the origin of eukaryotic cells (18–21), which has been a controversial issue.
A key question in the evolution of biological complexity (6, 7, 9, 12, 21, 22) is, how have integrated biological systems evolved? Darwinists (21, 23) proposed natural selection as the driving force of evolution. However, the striking similarities between biological and nonbiological complexities have led to the argument that a set of universal (or ahistorical) rules account for the formation of all complexities (22, 24, 25). The yeast protein interaction network is an example of a complex biological system and contributes to the complexity at the cellular level (26). By analyzing the growth pattern and reconstructing the evolutionary path of the yeast protein interaction network, we can address whether or not network growth is contingent on evolutionary history, which is the key disagreement between the Darwinian view and the universality view (22, 23, 27).
In this article, we studied how the yeast protein interaction network has evolved. We used graph theory to model the yeast protein interaction network. Each yeast protein is a node in the graph. Each pairwise interaction is a link between two nodes. Evolution of the yeast protein interaction network can then be inferred by analyzing the growth pattern of the graph. We classified all of the nodes (proteins) into isotemporal categories based on each protein's orthologous hits in several groups of genomes that are informative for yeast's evolutionary history. This scheme gives each protein a binary (b) value representing its evolutionary history. Proteins from the same isotemporal category share similar evolutionary histories. We then analyzed the interaction patterns within and between these isotemporal categories. Finally, we inferred the main path of the network evolution from six major isotemporal categories.
Materials and Methods
Data Collection. Genomic information of Saccharomyces cerevisiae was downloaded from the Saccharomyces Genome Database (ftp://genome-ftp.stanford.edu/pub/yeast/data_download) on August 13, 2002. Protein interaction data were obtained from the Comprehensive Yeast Genome Database at the Munich Information Center for Protein Sequences (MIPS) (http://mips.gsf.de/proj/yeast/CYGD/db/index.html) (28, 29) on May 28, 2002, and from the reliable subsets of data from high-throughput screens (30). We excluded self-interactions and those involving mitochondrion proteins. The combined data set contains 6,633 interaction pairs. Orthologous analyses of the annotated ORFs in the yeast genome were parsed out from the clusters of orthologous groups (COGs) of proteins (ftp://ftp.ncbi.nih.gov/pub/COG) (31, 32) and the published orthologous analysis from the Bork group at the European Molecular Biology Laboratory (EMBL) (30). Mitochondrion genes and a few inconsistent orthologous assignments were removed from the analysis.
Data Analysis. Protein interaction networks were treated as undirected graphs in adjacency list format (33). Permutations of the networks were carried out in the Chiba City Linux cluster in the Mathematics and Computer Science Division of Argonne National Laboratory (www.mcs.anl.gov/chiba). Presentation of the network was performed by the program pajek (http://vlado.fmf.uni-lj.si/pub/networks/pajek) (34). Distance matrix-based analyses were conducted in the r environment for statistical computing and graphics (www.r-project.org) (35). The neighbor-joining (NJ) tree was generated by paup* (http://paup.csit.fsu.edu) and presented by the program treeview (http://taxonomy.zoology.gla.ac.uk/rod/treeview.html) (36).
Statistical Analysis of Interaction and Traversal Patterns. To evaluate the interaction tendencies within and between isotemporal categories, we measured the deviation of each observed interaction frequency from its random expectation (37). The observed interaction frequency between categories i and j, 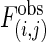 , is compared with the mean interaction frequency,
, is compared with the mean interaction frequency, 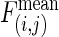 , of a series of null models in which all proteins have the same connectivities, but their interaction partners are randomly chosen (37) [termed the Maslov–Sneppen 2002 (MS02) null models]. To describe the deviations of the observed interaction frequencies from the random expectations, we used Z scores,
, of a series of null models in which all proteins have the same connectivities, but their interaction partners are randomly chosen (37) [termed the Maslov–Sneppen 2002 (MS02) null models]. To describe the deviations of the observed interaction frequencies from the random expectations, we used Z scores, 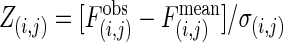 , where σ(i,j) is the SD of the interaction frequency between categories i and j in the MS02 null models.
, where σ(i,j) is the SD of the interaction frequency between categories i and j in the MS02 null models.
Similarly, we used the Z scores to measure the deviation of the average shortest path between two isotemporal categories from the mean of a series of isomorphic MS02 null models. This isomorphic MS02 null model retained the same topology with the original network. Network topology can greatly influence the average shortest path. The MS02 null model could change the total number of connected components in the original network and gave uninterpretable Z scores. The isomorphic null model was a simple method to exclude the topological influence on traversal path, and it enabled us to evaluate the association significance between two isotemporal categories.
Network Null Models. To generate an MS02 null model (37), the original network was first converted to pairwise-interacting nodes. These pairwise interacting nodes were then converted into an array of symbols. Permutation of this array of symbols was then used to generate a new list of pairwise-interacting nodes (self-pairing was prohibited during the permutation), which was then used to generate an MS02 null model in adjacency list format.
To generate an isomorphic MS02 null model, nodes with the same connectivity were concatenated into arrays of symbols. Permutation was then conducted on the arrays of symbols for each connectivity value. The original and the permutated arrays of symbols were then used to generate a lookup table in which each original node corresponded to a new node with the same connectivity. Based on this lookup table, all of the nodes in the original network were then replaced by the new nodes, resulting in a permutated network with the same topology.
Calculation of Average Shortest Path. We slightly modified the Dijkstra's algorithm to compute the shortest path (33). For a protein in isotemporal category i, its shortest path to isotemporal category j is defined as its traversal distance to the nearest neighbor in category j. The mean of the shortest paths to category j of all proteins in category i is taken as the distance from i to j, denoted as di→j. Distance from j to i, dj→i, is calculated similarly. The average shortest path between categories i and j is the average of di→j and dj→i.
Results
Isotemporal Classification of Proteins. To study the growth of the yeast protein interaction network, we classified all yeast proteins into isotemporal categories, based on the presence or absence of their orthologous hits in each of the six groups of the universal tree of life (38), namely hyperthermophilic eubacteria, other eubacteria (excluding the hyperthermophiles), euryarchaeota, crenarchaeota, fungi, and other eukaryotes (excluding fungi) (Fig. 1). The first four groups are evolutionary pivotal groups (19). The hyperthermophilic eubacteria and other eubacteria may reflect one of the earliest splits in the eubacterial domain (38–41). Likewise, crenarcheota and euryarchaeota represent an early split in the archaeal domain (19, 38). We separated the fungal genomes from other eukaryotes because they may reveal recent evolutionary changes of yeast. For the purpose of orthologous analysis, the yeast genome is excluded from the groups of fungi and other eukaryotes. We parsed out the orthologous hits from the COGs (31) and another published orthologous analysis (30). Because the proteins in each category share the same or similar evolutionary histories, these categories might have been added to the yeast genome at various temporal intervals during evolution, and can be considered as isotemporal categories.
Fig. 1.

Isotemporal classification of the yeast proteins. Isotemporal categories are designed through a binary (b) coding scheme. The b code represents the distribution of each yeast protein's orthologs in the universal tree of life. Bit value 1 indicates the presence of at least one orthologous hit for a yeast protein in a corresponding group of genomes, and bit value 0 indicates the absence of any orthologous hit. The presented example is 110011 in the b format and 51 in the d format. Orthologous identifications are based on COGs at the National Center for Biotechnology Information (31) and the results of the Bork group at the EMBL (30).
We designed a b coding scheme to represent the isotemporal categories (Fig. 1). The bits of the b coding scheme correspond to the six chosen evolutionary groups. For each yeast protein under study, the presence or absence of at least one orthologous hit in the genomes of each evolutionary group is represented by “1” or “0.” Mathematically, this six-bit coding scheme gives 64 categories, but the yeast genome contains 42 categories with nonrandom distributions because of evolutionary constraints (see Fig. 4, which is published as supporting information on the PNAS web site, www.pnas.org). For presentation convenience, we used both b codes and their decimal (d) values. For example, category b000011 is equivalent to category d3, which contains proteins whose orthologs are found in the groups of fungi and other eukaryotes.
Interaction Patterns in the Network. We constructed a credible protein interaction network by using the manually curated protein interaction pairs maintained at MIPS (28) and the reliable subsets of data from high-throughput screens (30). The generated protein interaction networks are treated as undirected graphs. We excluded all self-interactions because we analyzed the network growth from the perspective of node additions. For simplicity, we also excluded the mitochondrion-coded proteins. The generated network contains only 39 isotemporal categories, with a biased coverage favoring the well conserved proteins in categories b000011 and b111111 (see Fig. 5, which is published as supporting information on the PNAS web site). This bias may reflect the assumption that conserved proteins are functionally more important than nonconserved ones, and the former deserve more experimental effort (37). In addition, interactions between well conserved proteins can be confirmed by their orthologs in other species (30).
We used Z scores to evaluate the interaction significance within and between isotemporal categories, based on the MS02 null models (Fig. 2a). Positive Z scores indicate that observed interactions are more frequent than random expectations; negative Z scores indicate the opposite. Therefore, large positive Z scores indicate strong interaction tendencies, whereas large negative Z scores indicate that proteins in the two categories tend to avoid each other in the network. Because the protein interaction network is treated as an undirected graph, the matrix presentation of the Z scores of all categories is symmetric. The diagonal distribution of large positive Z scores indicates that yeast proteins tend to interact with proteins from the same or closely related isotemporal categories. The observed intracategory association tendencies are consistent with the intuitive notion that a new function likely requires a group of new proteins, and that the growth of the protein interaction network is under functional constraints. For example, category b000011 (d3) contains the eukaryote-conserved nodes with intracategory interaction tendency, Z(3, 3) = 7.1, indicating that nodes added during the eukaryotic expansion tend to interact among themselves. In addition, the preexisting network may also contain clusters constrained by function, and many of these clusters have been preserved during the network evolution. For example, category b111111 (d63) may contain the most ancient nodes, and Z(63, 63) = 13.6, which indicates that these nodes still tend to interact among themselves. The result here suggests that evolution of the yeast protein interaction network has undergone additions of clusters of nodes, which we term isotemporal clusters (detailed below).
Fig. 2.

Interaction patterns. (a) Z scores for all possible interactions of the isotemporal categories in the protein interaction network. For categories i and j, 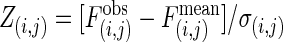 , where
, where 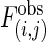 is the observed number of interactions,
is the observed number of interactions, 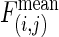 and σ(i,j) are the average number of interactions and the SD, respectively, in 10,000 MS02 null models (37). A cutoff value of 10 is chosen in this presentation. The data matrix is in Table 2, which is published as supporting information on the PNAS web site. (b) Z scores for the average shortest paths of the isotemporal categories in the largest component of the analyzed protein interaction network. For categories i and j,
and σ(i,j) are the average number of interactions and the SD, respectively, in 10,000 MS02 null models (37). A cutoff value of 10 is chosen in this presentation. The data matrix is in Table 2, which is published as supporting information on the PNAS web site. (b) Z scores for the average shortest paths of the isotemporal categories in the largest component of the analyzed protein interaction network. For categories i and j, 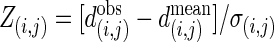 where
where 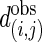 is the observed average shortest path,
is the observed average shortest path, 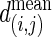 and σ(i,j) are the averaged average shortest path and the SD, respectively, in 500 isomorphic MS02 null models. A cutoff value of 5 is chosen in this presentation. The data matrix is in Table 3, which is published as supporting information on the PNAS web site.
and σ(i,j) are the averaged average shortest path and the SD, respectively, in 500 isomorphic MS02 null models. A cutoff value of 5 is chosen in this presentation. The data matrix is in Table 3, which is published as supporting information on the PNAS web site.
All observed negative Z scores are intercategorical. One of the most interesting ones is Z(3, 63) = –9.1, which indicates that the eukaryote-conserved proteins (b000011) tend to avoid the most conserved proteins (b111111).
To support the above conclusions, we also calculated the average shortest paths within and between the isotemporal categories in the largest connected component of the yeast protein interaction network. The above analysis considered only direct association, whereas the average shortest paths can measure indirect association. We used Z scores to evaluate traversal patterns within and between isotemporal categories, based on the isomorphic MS02 null models. Although this isomorphic null model is statistically overstringent, it is sufficient for evaluating the traversal profiles of the isotemporal categories. The Z score matrix shows that the intracategory traversal distances are usually significantly below random expectations (Fig. 2b). Thus, this analysis also shows that intracategory association tendencies are stronger than intercategory association tendencies.
Reconstruction of the Main Network Evolutionary Path. We reconstructed the main growth path of the network from the interaction patterns among the following six major isotemporal categories: b000000, b000001, b000011, b001111, b110011, and b111111. In our designed isotemporal categories, there are two groups of genomes for each domain of life (Eubacteria, Archaea, and Eukaryotes) (38). Categories b000011, b001111, b110011, and b111111 contain identical orthologous hits in both groups of genomes in each domain of life, and they are informative about the root of the universal tree of life (19, 38). Categories b000001 and b000000 may reveal the recent evolutionary history of the yeast. Furthermore, these six categories have large sample sizes.
We converted the Z score of intercategory interaction tendency into distance (dz) through a logit-like transformation, dz = 1/(1 + eZ), which transforms the Z scores into the range (0, 1). Positive Z scores correspond to small dz values because they indicate that the observed intercategory interactions are above random expectations. Conversely, negative Z scores correspond to large dz values. From the dz distance matrix, we inferred an NJ tree (42) that describes the intercategory interaction tendencies of the major isotemporal categories (Fig. 3a). This tree is essentially the blueprint that accounts for the expansion of the protein interaction network, by means of the addition of groups of proteins to the network at various periods during evolution. The main assembling order of the major groups is represented by the path from the ancient proteins (b111111) to eukaryote-conserved proteins (b000011) and then to recent proteins (b000001 and b000000). Assuming that there existed an ancestral protein interaction network represented by the b111111 nodes, and assuming that network evolution can be described by node additions, the path from the ancient proteins to the recent ones in the NJ tree would thereby describe the major path of the network growth.
Fig. 3.

The main path of network growth. (a) An NJ tree based on dz = 1/(1 + eZ), where Z is the Z score for interaction tendencies from Fig. 2 a. (b) An NJ tree based on dz′ = 1/(1 + e–Z), where Z is the Z score for the average shortest path from Fig. 2b. Both methods give the same branching pattern.
The positioning of b001111 (conserved between Archaea and Eukaryotes) and b110011 (conserved between Eubacteria and Eukaryotes) is consistent with the symbiotic hypothesis of the eukaryotic origin that argues for an archaeal host and a eubacterial symbiont (43).
Likewise, through the transformation, d′z = 1/(1+ e–Z), of the Z scores of the average shortest paths, we inferred an NJ tree with the same branching pattern (Fig. 3b). Therefore, by using two independent measurements, we observed that network evolution mirrors the universal tree of life.
Isotemporal Clusters in the Network. By using a single-linkage clustering method (44), we isolated the isotemporal clusters in the yeast protein interaction network by merging interacting proteins from the same isotemporal category into one node (see Fig. 6, which is published as supporting information on the PNAS web site). To estimate the clustering significance, we again used the isomorphic MS02 null model. For most isotemporal categories with relatively large populations, the numbers of their isotemporal clusters are significantly lower than the random expectations (Table 1). This result further supports the role of synergistic selection during network evolution. It is possible that new proteins are randomly added to the network. A single new addition to the network is more likely to be functionally irrelevant or deleterious, and tends to be filtered out during evolution, whereas additions of several interacting new proteins are more likely to be functional relevant and preserved. The observed isotemporal clusters and the proposed synergistic selection are consistent with the observed modularity in biological networks (7, 45).
Table 1. Numbers and sizes of major isotemporal clusters.
| Isotemporal categories |
Cluster numbers |
Average cluster sizes |
|||
|---|---|---|---|---|---|
| No. | Z score | P value | Size | Z score | |
| 000000 | 357 | -6.2 | <0.001 | 1.31 | 7.1 |
| 000001 | 272 | -2.6 | 0.007 | 1.42 | 2.8 |
| 000011 | 264 | -1.9 | 0.018 | 2.6 | 2.1 |
| 001111 | 46 | -7.4 | <0.001 | 2.13 | 10.9 |
| 110011 | 66 | -4.1 | <0.001 | 1.39 | 4.7 |
| 111111 | 199 | -4.2 | <0.001 | 1.67 | 4.9 |
Z scores and P values are calculated based on 1,000 isomorphic MS02 null models. A three-dimensional presentation of the isotemporal clusters is provided in Fig 6.
Discussion
Although we used the best annotated data available at the time of this study, the problems of false-positive and false-negative (14, 30, 46–50) data were not completely avoided. There is also the biased coverage toward conserved proteins (30). All these factors, however, likely affect the inter- and intracategory interactions randomly and so may not alter our main conclusions.
Our isotemporal classification of yeast proteins is limited by the sequence similarity search, the methods chosen to define orthologous groups, and the number of genomes available. These limitations, however, would largely affect the bits with 0 in the b coding scheme and would contribute to the large sample sizes of b000000 and b000001. Possibly, some b000011 proteins have been misclassified as b000001, and some b000001 proteins have been misclassified as b000000. As a result, some true b000011-b111111 associations may have been misclassified as b000001-b111111 or b000000-b111111. These misclassifications may affect both b000000 and b000001 to a similar extent and therefore may not drastically alter the inferred intercategory association tendencies among these categories. In addition, misclassification decreases intracategory Z scores, which means that the true intracategory association is actually more significant than estimated above.
The evolutionary origin of cellular life has been a controversial issue (18, 20, 51). The endosymbiotic hypothesis (19, 43) postulates an archaebaterium as the host and a eubacterium as the symbiont. From our observed significant intracategory association for all isotemporal categories of proteins, the significant separation tendency between b000011 (eucarya-conserved) and b111111 (ancient) proteins, and the inferred path of the network evolution, our result is strongly consistent with the endosymbiotic hypothesis. In addition, comparison of metabolic networks is also consistent with this hypothesis (5, 52).
The key disagreement between the Darwinian view and the universality view on the evolution of biological complexity is the role of historical contingency (22, 27). Undoubtedly, efforts to search for universal rules benefit our understanding on biological complexity. However, by using the yeast protein interaction network as an example, we observed a correlation between network evolution and the universal tree of life. This observation strongly argues that network evolution is not ahistorical, but is, in essence, a string of historical events.
Although the turnover rate of the protein interaction network is suggested to be very fast (9), our results suggest that many isotemporal clusters can still remain well preserved during evolution. The formation and conservation of isotemporal clusters during evolution may be the consequence of selection for the modular organization of the protein interaction network. The progressive nature of the network evolution and significant isotemporal clustering may have contributed to the hierarchical organization of modularity in biological networks in general (7). Because of the similarities between biological and nonbiological networks (1–3, 6, 7), isotemporal clustering and synergistic selection may be relevant in the evolution of many complex networks.
Supplementary Material
Acknowledgments
We thank Bill Martin, Kateryna Markova, Todd Oakley, Martin Feder, Xun Gu, Eugene Koonin, Natalia Maltsev, Maximino Aldana, Henrik Kaessmann, Leigh Van Valen, Thomas Nagylaki, Zhenglong Gu, Joel Bader, Anton Nekrutenko, Jianming Zhang, Geoffrey Morris, Tao Pan, Leo Kadanoff, Peter Bauman, Shinhan Shiu, Richard Blocker, and many others for assistance, suggestions, and discussions. This work was supported by National Institutes of Health Grants GM30998 and GM66104 (to W.-H.L.).
Abbreviations: MS02 null model, Maslov-Sneppen 2002 null model; NJ, neighbor-joining; b, binary; d, decimal.
References
- 1.Hartwell, L. H., Hopfield, J. J., Leibler, S. & Murray, A. W. (1999) Nature 402 C47–C52. [DOI] [PubMed] [Google Scholar]
- 2.Davidson, E. H., McClay, D. R. & Hood, L. (2003) Proc. Natl. Acad. Sci. USA 100 1475–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barabasi, A. L. (2002) Linked: The New Science of Networks (Perseus Publishing, Cambridge, MA).
- 4.Shen-Orr, S. S., Milo, R., Mangan, S. & Alon, U. (2002) Nat. Genet. 31 64–68. [DOI] [PubMed] [Google Scholar]
- 5.Podani, J., Oltvai, Z. N., Jeong, H., Tombor, B., Barabasi, A. L. & Szathmary, E. (2001) Nat. Genet. 29 54–56. [DOI] [PubMed] [Google Scholar]
- 6.Milo, R., Shen-Orr, S., Itzkovitz, S., Kashtan, N., Chklovskii, D. & Alon, U. (2002) Science 298 824–827. [DOI] [PubMed] [Google Scholar]
- 7.Ravasz, E., Somera, A. L., Mongru, D. A., Oltvai, Z. N. & Barabasi, A. L. (2002) Science 297 1551–1555. [DOI] [PubMed] [Google Scholar]
- 8.Wolf, Y. I., Karev, G. & Koonin, E. V. (2002) BioEssays 24 105–109. [DOI] [PubMed] [Google Scholar]
- 9.Wagner, A. (2001) Mol. Biol. Evol. 18 1283–1292. [DOI] [PubMed] [Google Scholar]
- 10.Fraser, H. B., Hirsh, A. E., Steinmetz, L. M., Scharfe, C. & Feldman, M. W. (2002) Science 296 750–752. [DOI] [PubMed] [Google Scholar]
- 11.Rzhetsky, A. & Gomez, S. M. (2001) Bioinformatics 17 988–996. [DOI] [PubMed] [Google Scholar]
- 12.Wagner, A. (2003) Proc. R. Soc. London Ser. B 270 457–466. [Google Scholar]
- 13.Uetz, P., Giot, L., Cagney, G., Mansfield, T. A., Judson, R. S., Knight, J. R., Lockshon, D., Narayan, V., Srinivasan, M., Pochart, P., et al. (2000) Nature 403 623–627. [DOI] [PubMed] [Google Scholar]
- 14.Ito, T., Chiba, T., Ozawa, R., Yoshida, M., Hattori, M. & Sakaki, Y. (2001) Proc. Natl. Acad. Sci. USA 98 4569–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ito, T., Tashiro, K., Muta, S., Ozawa, R., Chiba, T., Nishizawa, M., Yamamoto, K., Kuhara, S. & Sakaki, Y. (2000) Proc. Natl. Acad. Sci. USA 97 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tong, A. H., Drees, B., Nardelli, G., Bader, G. D., Brannetti, B., Castagnoli, L., Evangelista, M., Ferracuti, S., Nelson, B., Paoluzi, S., et al. (2002) Science 295 321–324. [DOI] [PubMed] [Google Scholar]
- 17.Tong, A. H., Evangelista, M., Parsons, A. B., Xu, H., Bader, G. D., Page, N., Robinson, M., Raghibizadeh, S., Hogue, C. W., Bussey, H., et al. (2001) Science 294 2364–2368. [DOI] [PubMed] [Google Scholar]
- 18.Woese, C. R. (2002) Proc. Natl. Acad. Sci. USA 99 8742–8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown, J. R. & Doolittle, W. F. (1997) Microbiol. Mol. Biol. Rev. 61 456–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin, W. & Russell, M. J. (2003) Philos. Trans. R. Soc. London B 358 59–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maynard Smith, J. & Szathmáry, E. (1995) The Major Transitions in Evolution (W. H. Freeman Spektrum, Oxford).
- 22.Kauffman, S. (1993) The Origins of Order: Self-organization and Selection in Evolution (Oxford Univ. Press, New York).
- 23.Corning, P. A. (1995) Syst. Res. 12 89–121. [Google Scholar]
- 24.Thompson, D. W. (1917) On Growth and Form (Cambridge Univ. Press, Cambridge, U.K.).
- 25.Wolfram, S. (2002) A New Kind of Science (Wolfram Media, Champaign, IL).
- 26.Oltvai, Z. N. & Barabasi, A. L. (2002) Science 298 763–764. [DOI] [PubMed] [Google Scholar]
- 27.Gould, S. J. (2002) The Structure of Evolutionary Theory (Harvard Univ. Press, Cambridge, MA).
- 28.Mewes, H. W., Frishman, D., Guldener, U., Mannhaupt, G., Mayer, K., Mokrejs, M., Morgenstern, B., Munsterkotter, M., Rudd, S. & Weil, B. (2002) Nucleic Acids Res. 30 31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mewes, H. W., Albermann, K., Heumann, K., Liebl, S. & Pfeiffer, F. (1997) Nucleic Acids Res. 25 28–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Mering, C., Krause, R., Snel, B., Cornell, M., Oliver, S. G., Fields, S. & Bork, P. (2002) Nature 417 399–403. [DOI] [PubMed] [Google Scholar]
- 31.Tatusov, R. L., Koonin, E. V. & Lipman, D. J. (1997) Science 278 631–637. [DOI] [PubMed] [Google Scholar]
- 32.Tatusov, R. L., Natale, D. A., Garkavtsev, I. V., Tatusova, T. A., Shankavaram, U. T., Rao, B. S., Kiryutin, B., Galperin, M. Y., Fedorova, N. D. & Koonin, E. V. (2001) Nucleic Acids Res. 29 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cormen, T. H., Leiserson, C. E. & Rivest, R. L. (1990) Introduction to Algorithms (MIT Press, Cambridge, MA).
- 34.Batagelj, A. & Mrvar, A. (1998) Connections 21 47–57. [Google Scholar]
- 35.Ripley, B. D. (2001) MSOR Connections 1 23–25. [Google Scholar]
- 36.Page, R. D. M. (1996) Comput. Appl. Biosci. 12 357–358. [DOI] [PubMed] [Google Scholar]
- 37.Maslov, S. & Sneppen, K. (2002) Science 296 910–913. [DOI] [PubMed] [Google Scholar]
- 38.Woese, C. R. (1987) Microbiol. Rev. 51 221–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Achenbach-Richter, L., Gupta, R., Stetter, K. O. & Woese, C. R. (1987) Syst. Appl. Microbiol. 9 34–39. [DOI] [PubMed] [Google Scholar]
- 40.Deckert, G., Warren, P. V., Gaasterland, T., Young, W. G., Lenox, A. L., Graham, D. E., Overbeek, R., Snead, M. A., Keller, M., Aujay, M., et al. (1998) Nature 392 353–358. [DOI] [PubMed] [Google Scholar]
- 41.Nelson, K. E., Clayton, R. A., Gill, S. R., Gwinn, M. L., Dodson, R. J., Haft, D. H., Hickey, E. K., Peterson, J. D., Nelson, W. C., Ketchum, K. A., et al. (1999) Nature 399 323–329. [DOI] [PubMed] [Google Scholar]
- 42.Saitou, N. & Nei, M. (1987) Mol. Biol. Evol. 4 406–425. [DOI] [PubMed] [Google Scholar]
- 43.Martin, W., Hoffmeister, M., Rotte, C. & Henze, K. (2001) Biol. Chem. 382 1521–1539. [DOI] [PubMed] [Google Scholar]
- 44.Anderberg, M. R. (1973) Cluster Analysis for Applications (Academic, New York).
- 45.Rives, A. W. & Galitski, T. (2003) Proc. Natl. Acad. Sci. USA 100 1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deane, C. M., Salwinski, L., Xenarios, I. & Eisenberg, D. (2002) Mol. Cell Proteomics 1 349–356. [DOI] [PubMed] [Google Scholar]
- 47.Edwards, A. M., Kus, B., Jansen, R., Greenbaum, D., Greenblatt, J. & Gerstein, M. (2002) Trends Genet. 18 529–536. [DOI] [PubMed] [Google Scholar]
- 48.Bader, G. D. & Hogue, C. W. (2002) Nat. Biotechnol. 20 991–997. [DOI] [PubMed] [Google Scholar]
- 49.Aloy, P. & Russell, R. B. (2002) FEBS Lett. 530 253–254. [DOI] [PubMed] [Google Scholar]
- 50.Kemmeren, P., van Berkum, N. L., Vilo, J., Bijma, T., Donders, R., Brazma, A. & Holstege, F. C. (2002) Mol. Cell 9 1133–1143. [DOI] [PubMed] [Google Scholar]
- 51.Brenner, S. (1998) Science 282 1411–1412. [Google Scholar]
- 52.Jeong, H., Tombor, B., Albert, R., Oltvai, Z. N. & Barabasi, A. L. (2000) Nature 407 651–654. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}