

Abstract
The exceptional sensitivity of Mycobacterium tuberculosis to isonicotinic acid hydrazide (INH) lacks satisfactory definition. M. tuberculosis is a natural mutant in oxyR, a central regulator of peroxide stress response. The ahpC gene, which encodes a critical subunit of alkyl hydroperoxide reductase, is one of the targets usually controlled by oxyR in bacteria. Unlike in mycobacterial species less susceptible to INH, the expression of ahpC was below detection limits at the protein level in INH-sensitive M. tuberculosis and Mycobacterium bovis strains. In contrast, AhpC was detected in several series of isogenic INH-resistant (INHr) derivatives. In a demonstration of the critical role of ahpC in sensitivity to INH, insertional inactivation of ahpC on the chromosome of Mycobacterium smegmatis, a species naturally insensitive to INH, dramatically increased its susceptibility to this compound. These findings suggest that AhpC counteracts the action of INH and that the levels of its expression may govern the intrinsic susceptibility of mycobacteria to this front-line antituberculosis drug.
Keywords: oxidative stress, ahpC, oxyR, gene replacements, mycobacteria
Tuberculosis has never ceased to be a global health problem (1). Much of the recent attention that this disease has received can be attributed to the resurgence of tuberculosis in industrialized countries. This is further compounded by the emergence of drug-resistant strains of the etiologic agent Mycobacterium tuberculosis, including variants recalcitrant to treatments with the front-line antituberculosis agent isonicotinic acid hydrazide (isoniazid; INH) (1). Since its introduction in 1952 (2) as a potent agent for treatment of tuberculosis, INH has proven to be an invaluable therapeutic agent. However, despite numerous proposals (3–5) and recent developments (6, 7), the effects of this compound on the mycobacterial cell still lack a complete definition.
The initial advances in understanding the antimycobacterial action of INH came from the explorations of the mechanisms of the emergence of INH resistance in M. tuberculosis (8). Based on recent genetic analyses, it has been widely accepted that katG, a gene encoding catalase peroxidase, is a major site of mutations conferring INH resistance on M. tuberculosis (9, 10). These analyses have led to the current model in which INH is activated by a peroxidatic reaction in the presence of KatG (3, 5, 11). The activated product of INH is believed to act on InhA (6, 7, 12), an enzyme implicated in mycolic acid synthesis (6). In addition to the proposed action of INH on InhA and other putative targets (5, 6), the intracellular metabolism of INH is known to generate reactive oxygen intermediates (11, 13–16) that may have deleterious effects on mycobacterial cells.
While the majority of current investigations have been aimed at understanding the emergence of INH-resistant (INHr) strains, we have recently addressed the fundamental question concerning the mechanisms underlying the natural hypersensitivity of M. tuberculosis to INH (17, 18). This organism stands out among mycobacteria as being several orders of magnitude more sensitive to INH than the majority of other species (3, 5). In efforts to investigate this phenomenon, we have recently shown (17) that all strains of M. tuberculosis and other members of the M. tuberculosis complex (Mycobacterium africanum, Mycobacterium bovis, and Mycobacterium microti) are natural mutants in oxyR, the putative M. tuberculosis equivalent of the central regulator of peroxide stress response in enteric bacteria. Similar results have been reported by others (19). The sequence of the oxyR region and the lesions inactivating the oxyR gene are identical in all type strains of the M. tuberculosis complex, which also share high susceptibility to INH (17).
In Enterobacteriaceae, oxyR activates specific defense mechanisms that detoxify reactive oxygen intermediates and remove their harmful products (20, 21). One of the genes controlled by oxyR in enteric bacteria is ahpC, encoding the subunit of alkyl hydroperoxide reductase that reduces organic peroxides to their corresponding alcohols (20). Significantly, ahpC and oxyR are tightly linked and divergently transcribed in M. tuberculosis (Fig. 1A) and in the majority of other mycobacteria studied (17). Point mutations in the oxyR–ahpC intergenic region have been noted in some strains of M. tuberculosis, suggesting their potential role in the emergence of INHr variants (18, 22, 24). It has been proposed that the putative increase in AhpC in such mutants could either directly counteract INH effects (18, 22) or could act as a compensatory change enhancing the viability of katG mutants (18, 24). Since harmful oxygen by-products of INH metabolism have been implicated in the toxicity of INH in bacteria (5, 11, 13–16), we investigated herein whether ahpC plays a role in the intrinsic sensitivity of mycobacteria to INH. We demonstrate that ahpC expression is a critical factor contributing to the differential susceptibility of mycobacteria to INH.
Figure 1.
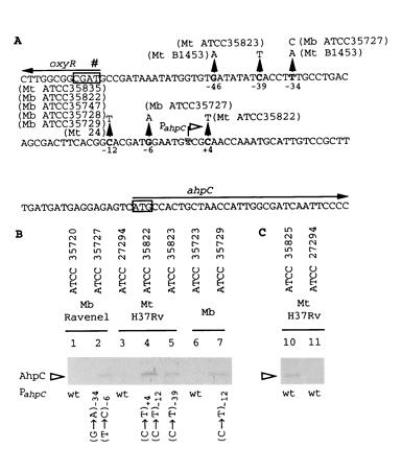
(A) DNA sequence of the oxyR–ahpC intergenic region from M. tuberculosis and M. bovis. The previously reported (17, 22) and additional mutations are indicated by upward pointing triangles. Positions of the nucleotide substitutions are relative to the ahpC mRNA start site (Fig. 2). Strain designations are given next to the corresponding mutations. Boxed sequences, start codon of ahpC and the destroyed start codon (#) of oxyR (17). Arrows, direction of transcription. (B) Western blot analysis of AhpC production in three series of INHr derivatives that carry ahpC promoter mutations in addition to katG lesions. Lanes: 2, 4, 5, 7, INHr derivatives; 1, 3, and 6, parental INHs strains. (C) AhpC production in an INHr derivative (lane 10) of M. tuberculosis H37Rv (lane 11) that does not carry the ahpC promoter alterations. Anti-DirA antibody that recognizes mycobacterial AhpC (18, 23) was used for Western blot analysis. The strains tested are indicated above the blot and the corresponding ahpC promoter mutations are indicated below the blot. Mb, M. bovis; Mt, M. tuberculosis; wt, wild type.
MATERIALS AND METHODS
Bacterial Strains, Plasmids, and Growth Conditions.
All M. tuberculosis and M. bovis strains were from the American Type Culture Collection (ATCC). Mycobacterium smegmatis mc2 155 has been described (25). The strains VD1865-6 and VD1865-38 were two independently generated ahpC::Kmr mutants of M. smegmatis (where Kmr is kanamycin resistant). Mycobacteria were grown in Middlebrook 7H9 medium or on 7H10 plates (Difco) supplemented with 0.05% Tween 80 and ADC enrichment for M. smegmatis or OADC for M. tuberculosis. All manipulations of live M. tuberculosis were carried out under Biosafty Level 3 conditions. M. tuberculosis was inactivated by heating at 80°C for 1 h.
Recombinant DNA Techniques, Genetic Methods, and Allelic Replacements.
To generate the fragment used for inactivation of ahpC on the M. smegmatis chromosome via homologous recombination, a 1.2-kb SpeI–NheI Kmr cassette from pMV206 was inserted into the BspEI site of the M. smegmatis ahpC gene on pDP81 (18), resulting in pDP83 with a 2.5-kb PstI fragment carrying the ahpC::Kmr construct. This fragment was purified and introduced into M. smegmatis mc2 155 by electroporation (26). Potential recombinants were selected by plating cells on 7H10 medium supplemented with 0.2% pyruvate and kanamycin at 10 μg/ml, colonies were screened for homologous recombination events by PCR, and allelic replacements were confirmed by Southern blot analysis in candidate isolates. No significant differences in catalase activities [the rate of A240 decrease was 0.04 ± 0.02 unit versus 0.06 ± 0.02 unit in the standard Beers–Sizer assay (27) using 25 μg of crude protein extracts] or in growth rates were observed between the parental ahpC+ strain and its ahpC::Kmr derivatives.
DNA Amplification and Sequencing.
The oxyR–ahpC intergenic region was amplified by PCR from M. tuberculosis and M. bovis using the previously described (17) oligonucleotides Ahp1P (5′-GCTTGATGTCCGAGAGCATCG-3′; positions +347 to +327 relative to the ATG of ahpC) and OxyR4 (5′-GGTCGCGTAGGCAGTGCCCC-3′; positions −355 to −336 relative to the ahpC initiation codon). The nucleotide sequences of the oxyR–ahpC intergenic region (positions +33 to −117, relative to the initiation codon of ahpC) was determined directly on the PCR products employing standard PCR DNA sequencing methods (28).
RNA Isolation and S1 Nuclease Protection Analysis.
Total cellular RNA was isolated by centrifugation through a cushion of 5.7 M CsCl as described (18). Single-stranded hybridization probes for ahpC were prepared using plasmid pVDtb#3 (17, 18) with the M. tuberculosis oxyR–ahpC region on a 5.5-kb BamHI genomic insert. The ahpC-specific oligonucleotide Ahp4 (5′-GGTGAAGTAGTCGCCGGGCT-3′; positions +108 to +89, relative to the ahpC initiation codon) was used to generate a uniformly labeled (32P) single-stranded probe and to produce the corresponding sequencing ladder as described (18). Equal amounts of RNA (33 μg) were hybridized with aliquots of the radioactively labeled probe. S1 nuclease protection analysis was carried out as described (18). S1 nuclease digestion products were analyzed on sequencing gels (7.5% polyacrylamide/8 M urea/100 mM Tris/100 mM boric acid/2 mM EDTA, pH 8.3) along with the sequencing ladder. Because of the uniform labeling of single-stranded DNA probes, which dramatically improves the sensitivity of the assay, radioactive decay contributes to the presence of multiple bands corresponding to the 5′ end of mRNA, as has been noted (18).
Immunoblot Analysis.
M. smegmatis, M. tuberculosis, and M. bovis were grown to midlogarithmic phase and crude protein extracts were obtained by homogenization in a Mini Beadbeater (Biospec Products, Bartlesville, OK) for 2 min at 2800 rpm. The cellular debris and beads were removed by centrifugation and the supernatant was mixed with an equal volume of 2× SDS/PAGE sample loading buffer and analyzed on SDS/11% polyacrylamide gels. The proteins were transferred onto Immobilon-P membranes (Millipore) by electroblotting and subjected to Western blot analysis using rabbit antiserum to DirA (AhpC) of Corynebacterium diphtheriae (23) that recognizes mycobacterial AhpC (18).
INH Sensitivity Determination.
A previously reported (29) disk INH-inhibition assay was used with some modifications. M. smegmatis strains mc2155 (ahpC+) and its ahpC::Kmr derivatives VD1865-6 and VD1865-38, grown to equal densities, were mixed with soft 7H10 agar and plated. Paper discs (6 mm in diameter) were impregnated with 10 μl of INH (1 mg/ml) and placed on top of the solidified soft agar. Zones of growth inhibition were measured after overnight incubation at 37°C.
RESULTS
Mapping of the ahpC mRNA Start Site and Detection of Transcription in M. bovis. The occurrence of point mutations in the oxyR–ahpC intergenic region in M.
tuberculosis and M. bovis has been reported (18, 22). These mutations have been observed in some INHr strains, and a particularly high incidence has been noted in ΔkatG mutant isolates (18) as summarized in Fig. 1A. It has been proposed that such mutations may cause increased ahpC transcription thus contributing to the emergence of INHr strains by either compensating for the loss of katG activity (18) or directly contributing to the low-level INH resistance (18, 22). To investigate whether mutations in the putative promoter region of ahpC result in detectable ahpC transcription, we first mapped the promoter for M. bovis ahpC using S1 nuclease protection analysis and RNA extracted from M. bovis bacillus Calmette–Guérin (BCG) Montreal ATCC35735 and its derivative ATCC35747, which carries the most common change in the oxyR–ahpC region (C12 → T12) (Fig. 1A). The expectation was that the transcript, if any, would be observed in the ahpC promoter mutant. The results of these experiments (Fig. 2) can be summarized as follows: (i) The ahpC mRNA 5′ end in M. bovis was located within the ahpC–oxyR intergenic region and was 42 bp upstream of the start codon of ahpC (Fig. 1A and 2). (ii) This finding positioned the most frequent promoter mutation (Fig. 1A) at position −12 relative to the mRNA start site. (iii) As anticipated, increased transcription was observed in the mutant derivative (Fig. 2, lane 2). (iv) Somewhat surprisingly, significant expression of ahpC was also observed (Fig. 2A, lane 1) in the parental strain M. bovis BCG ATCC35735 carrying the wild-type ahpC promoter.
Figure 2.
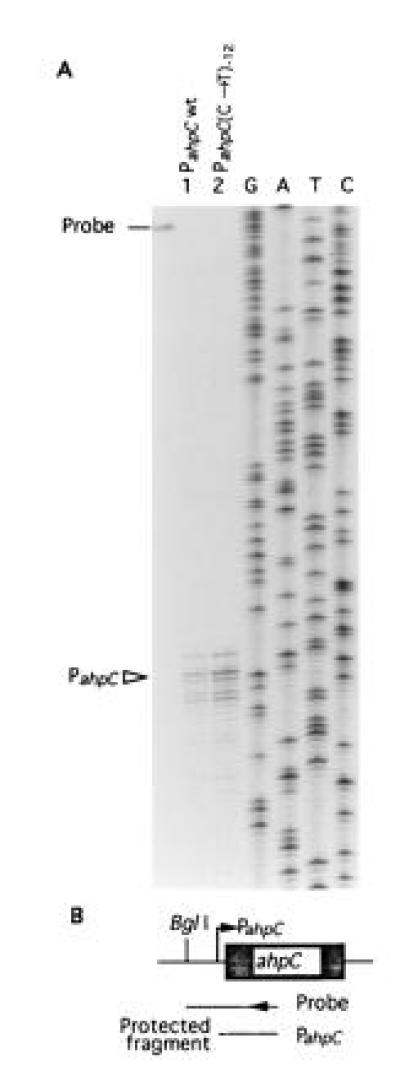
S1 nuclease protection mapping of the ahpC mRNA 5′ end in M. bovis. (A) RNA was isolated from M. bovis bacillus Calmette–Guérin (BCG) ATCC35735 (lane 1) and M. bovis BCG ATCC35747 (lane 2). These strains have been examined for AhpC production (18) and displayed same relationships as shown in Fig. 1. Bar, location of the untreated probe. (B) Schematic representation of the probe and protected fragment in relationship to ahpC and its upstream region. Single-stranded S1 nuclease probe was generated using the same primer that served to generate DNA sequencing ladder (GATC). BglI site was the other end of the probe. Triangle (PahpC), ahpC mRNA 5′ end; wt, wild type; (C → T)−12, C → T transition at position −12 relative to the mRNA start site.
Increased AhpC Production in M. tuberculosis with ahpC Promoter Mutations.
We investigated ahpC expression at the protein level in three series of isogenic strains: (i) the INHs parent M. tuberculosis H37Rv (ATCC27294) and its INHr derivatives ATCC35822 [carrying a double ahpC promoter mutation at positions −12 and +4 (Fig. 1A) and a full-length katG deletion which is considered to be the cause of its high INH resistance (22)] and ATCC35823 [carrying an ahpC promoter mutation at position −39 (GenBank accession no. U57761U57761; Fig. 1A) and a missense T274 → P274 mutation that renders KatG inactive (30)]; (ii) M. bovis 35723 (INHs) and its INHr derivative ATCC 35729 [with a typical C12 to T12 transition in the ahpC promoter (22) and a nonsense mutation W198 → Stop in katG (30)]; and (iii) M. bovis Ravenel ATCC35720 (INHs) and its INHr derivative ATCC35727 [carrying a double ahpC promoter mutation at positions −34 and −6 (Fig. 4A) and a 76-bp deletion in katG 106 bp downstream of the katG initiation codon (30)]. Western blot analyses with the antibody against DirA (AhpC) of C. diphtheriae (23), which recognizes mycobacterial AhpC (18), indicated that AhpC was observed only in the mutant INHr progeny while AhpC was not detectable in any of the corresponding parental INHs strains (Fig. 1B). These observations are consistent with the activation of ahpC expression in ahpC promoter mutants resulting in detectable production of AhpC.
Figure 4.
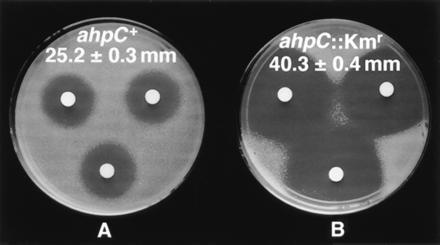
Inactivation of ahpC causes hypersensitivity to INH in M. smegmatis. Shown are growth inhibition zones with INH in M. smegmatis mc2 155 (ahpC+) and M. smegmatis VD1865-6 (ahpC::Kmr). Discs were impregnated with 10 μl of INH at 1 mg/ml and placed on top of the soft agar with corresponding strains. (A), M. smegmatis mc2 155; (B), M. smegmatis VD1865-6.
Multiple Mechanisms Lead to Enhanced AhpC Production in M. tuberculosis.
While testing the series of strains described above, we also examined another isogenic INHr derivative of M. tuberculosis H37Rv, strain ATCC35825, that has an enzymatically functional catalase peroxidase (30). This strain displays an intermediate level of INH resistance [5 μg/ml (30)] and carries the commonly observed Arg463 → Leu463 polymorphism, considered by some researchers to confer low-level resistance to INH (10, 30–33). However, this polymorphism is present in all M. bovis and M. microti strains (10) and does not affect catalase or peroxidase activity (30, 32); it has also been reported in a number of M. tuberculosis strains showing normal INH sensitivity (10, 34, 35). When tested for ahpC promoter mutations, M. tuberculosis ATCC35825 did not carry any changes in the oxyR–ahpC intergenic region. Surprisingly, we could detect AhpC by immunoblot analysis in this variant while it was absent in the parental strain H37Rv (Fig. 1C). These results indicate that AhpC production is increased in at least some INHr isolates of M. tuberculosis that do not carry mutations in the ahpC promoter region. These observations suggest the existence of multiple mechanisms, besides ahpC promoter mutations, that can lead to enhanced production of AhpC with implications for the emergence of INH resistance in M. tuberculosis.
Inactivation of ahpC in M. smegmatis Causes Hypersensitivity to INH.
In addition to INH-sensitive (INHs) strains of M. tuberculosis, AhpC is also not detectable (18) in Mycobacterium aurum, a fast growing species highly susceptible to INH (18, 36). In contrast, AhpC is produced in significant amounts in M. smegmatis, an organism epitomizing mycobacteria with low sensitivity to INH (3, 5, 9). This suggested to us that the differences in ahpC expression may correlate with the intrinsic sensitivity to INH in mycobacterial species. To test this hypothesis, we insertionally inactivated the recently cloned and characterized ahpC gene (18) on the chromosome of M. smegmatis and examined INH sensitivity of the resulting strains (Fig. 3A). Two independent ahpC::Kmr mutant isolates were obtained (VD1865-6 and VD1865-38). The gene replacements were confirmed by Southern blot analysis (Fig. 3B). The mutant ahpC::Kmr strains no longer produced AhpC as determined by Western blot analysis (Fig. 3C). The inactivation of ahpC caused a striking increase in susceptibility of M. smegmatis to INH as illustrated in Fig. 4. While the wild-type M. smegmatis showed a growth inhibition zone of 25.2 ± 0.3 mm, its ahpC::Kmr derivatives displayed an inhibition zone of 40.3 ± 0.4 mm [P value (t test) was 9 × 10−17]. Depending upon growth conditions, minimal inhibitory concentration of INH was decreased 4-fold to one order of magnitude for the mutant strain. The effect was specific for INH since the ahpC::Kmr derivatives did not show significant differences in sensitivity to other drugs such as rifampicin [23.3 ± 0.9 mm for ahpC+ versus 24.0 ± 0.6 mm for ahpC::Kmr cells; P value (t test) was 0.6] in disk inhibition assays. These results demonstrate that AhpC contributes to the relative ability of mycobacteria to resist the action of INH.
Figure 3.
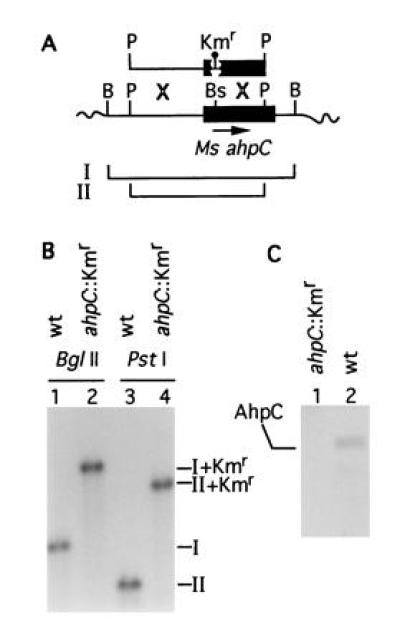
Inactivation of ahpC on the chromosome of M. smegmatis. (A) Schematic representation of the recombinational events between chromosomal ahpC and ahpC::Kmr on a 2.5-kb PstI fragment introduced into M. smegmatis mc2 155 by electroporation. B, BglII; P, PstI; Bs, BspEI. Balloon, 1.2-kb Kmr cassette. Lines I and II correspond to the wild-type BglII and PstI fragments, respectively. (B) Southern blot hybridization analysis of the insertional inactivation of ahpC in the strain VD1865-6. Lanes: 1 and 3, M. smegmatis mc2 155 ahpC+; 2 and 4, M. smegmatis VD1865-6. I, 1.7-kb BglII fragment (wt); II, 1.3-kb PstI fragment (wt). I+Kmr (2.9 kb) and II+Kmr (2.5 kb) are the corresponding bands hybridizing with the ahpC probe in the strain VD1865-6. (C) Western blot analysis of AhpC expression in the parent strain M. smegmatis mc2 155 (wt) and its mutant derivative VD1865-6 (ahpC::Kmr). Equal amounts of protein from crude extracts separated by SDS/PAGE were probed with anti-DirA antibody.
DISCUSSION
In this work, we have demonstrated that ahpC plays a role in the intrinsic sensitivity of mycobacteria to INH. This was accomplished by insertional inactivation of the ahpC gene in M. smegmatis, which increased the susceptibility of this organism to INH. In addition, the presented data indicate that the gene product of ahpC is below detection limits in all INHs strains of M. tuberculosis and M. bovis tested but that its transcription is increased and its gene product can be detected by immunoblots in M. tuberculosis isolates with mutations in the ahpC promoter. Furthermore, mechanisms besides ahpC promoter mutation appear to enhance AhpC production or stability in some M. tuberculosis strains with low-level INH resistance. These findings, together with the previous demonstration of a reduced susceptibility of M. tuberculosis to INH upon introduction of cosmids carrying the complete oxyR and ahpC genes of Mycobacterium leprae (17), support the notion that the defect in oxidative stress response in the tubercle bacillus is a major contributing factor to its exceptionally high INH sensitivity.
Mycobacteria show a wide range of innate susceptibilities to INH (3, 5). At one extreme are the highly sensitive members of the M. tuberculosis complex, while at the other are organisms such as M. smegmatis that are affected only by high concentrations of INH (3, 5, 9). Within the group of INHs organisms is Mycobacterium aurum, a fast growing nonpathogenic species that approaches INH susceptibility levels in M. tuberculosis (18, 36). In a recent analysis, AhpC could not be detected in M. aurum, in further support of the inverse correlation between ahpC expression and INH susceptibility (18). Since peroxidatic activation of INH involves production of damaging reactive oxygen intermediates (3, 11, 15, 16), it is conceivable that AhpC may counteract these effects by reducing oxidized targets or by detoxifying the activated INH. In this model, organisms with no or less AhpC may be at a disadvantage when exposed to INH.
The dysfunction of the peroxide stress response, which can be at least partially attributed to the lesions in oxyR (17), appears to be a major underlying cause of the exceptional sensitivity of M. tuberculosis to INH. Since ahpC is most likely only one of the genes controlled by oxyR [e.g., besides AhpC, eight other polypeptides inducible by peroxides have been identified in M. smegmatis (18)], additional putative members of the regulon may also participate in these processes. It will be of interest to determine whether such putative elements display cumulative effects in protection against INH. It is also possible that other previously appreciated peculiarities of M. tuberculosis, recently reviewed by Zhang and Young (5), may contribute to the overall sensitivity levels. However, such notions must await experimental support. For example, it has been speculated that the absence of the second catalase (katE) may be the reason for high susceptibility of M. tuberculosis to INH. However, it has been recently shown (37) that introduction of the second catalase (katE) in M. tuberculosis did not lower its sensitivity to INH. Although mycolic acid biosynthesis is a likely target for INH (6, 7, 12), this aspect of mycobacterial physiology alone may not suffice to explain the dramatic differences in sensitivities to INH. InhA, one of the suspected specific targets for INH (6, 7, 12), is also present in M. smegmatis (6) and is a close homolog of a pyridine nucleotide-linked enoyl reductase involved in the biogenesis of fatty acids in Escherichia coli (38), attesting to the ubiquitous presence of these factors. As an added curiosity and in keeping with our results, inactivation of oxyR and ahpC can render enteric bacteria, normally insensitive to INH, susceptible to this drug (29).
While the primary objective of our study, a relationship between ahpC activity and the intrinsic sensitivity to INH in several mycobacterial species, has been established, the role of ahpC in the emergence of INH resistance in M. tuberculosis is at present a controversial issue. Based on the effects that ahpC expression has on mycobacterial susceptibility to INH (18, 22), it appears only reasonable to expect that upregulation of ahpC could decrease sensitivity of M. tuberculosis to this drug. However, the demonstration of the role of such putative processes is complicated by the high incidence of ahpC promoter mutations in strains that already carry a mutation in katG (18). One interpretation is that AhpC compensates for the lack of katG expression in such isolates; alterations increasing ahpC expression may simply represent second-site suppressor mutation in strains that display high levels of INH resistance due to the loss of KatG (18). However, activation of ahpC may alone confer low-level resistance to INH since complementation of katG in some strains with dual katG and ahpC promoter mutations does not completely restore the INH sensitivity (22). In a recent study involving a large collection of clinical isolates that will be reported separately (S. Sreevantsan, Y.S., V.D., and J. M. Musser, unpublished data), a significant number of INHr strains have been identified carrying alterations only in the ahpC promoter. Out of 20 INHr clinical isolates carrying mutations in the ahpC promoter region at various positions between −45 and −4 relative to the ahpC mRNA start site, six strains had wild-type katG and inhA alleles while an additional group of six isolates had either the common R463L polymorphism or a neutral change in katG along with an intact inhA gene. In addition, a smaller subset of INHr strains carried missense mutation in the structural portion of the ahpC gene. These findings appear to support an independent role for ahpC mutations in the emergence of low level INH resistance in M. tuberculosis. However, due to the difficulty in demonstrating increased resistance to INH in M. tuberculosis harboring plasmid-borne ahpC (24), this issue remains open for further investigation.
It will also be of interest to determine the nature of processes causing increased AhpC levels in some strains that do not carry mutations in the ahpC promoter and whether similar mechanisms can operate in some clinical isolates with low-level INH resistance. Perhaps additional parallels concerning the regulation of oxidative stress response exist between mycobacteria and other bacteria. In Enterobacteriaceae, this system appears to include elements of posttranslational control (21) and in C. diphtheriae involves iron regulation (23). Further investigations of these putative regulatory mechanisms and complementary studies aimed at understanding potential consequences of the absence of oxyR function in M. tuberculosis on the course of infection and pathogenesis in tuberculosis may also shed light on host–pathogen interactions that lead to the fascinating phenomenon of the loss of oxyR.
Acknowledgments
We thank S. S. Tai for anti-DirA antibody and M. Schurr, L. Via, and H. Yu for their comments. This work was supported by Grant AI35217 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Abbreviations: ATCC, American Type Culture Collection; INH, isonicotinic acid hydrazide (isoniazid); INHr and INHs, INH-resistant and -sensitive, respectively; Kmr, kanamycin-resistant.
Data deposition: The sequences reported in this paper have been deposited in the GenBank data base (accession nos. U57977U57977 and U57760U57760 for M. tuberculosis H37Ra ATCC25177 and its derivative ATCC35835, respectively; U57761U57761 and U58030U58030 for the M. tuberculosis H37Rv derivatives ATCC35823 and ATCC35825, respectively; U57978U57978 and U57762U57762 for M. bovis Ravenel ATCC35720 and its derivative ATCC35727, respectively; and U58031U58031 for M. bovis ATCC35728).
References
- 1.Bloom B R, Murray C J L. Science. 1994;257:1055–1064. doi: 10.1126/science.257.5073.1055. [DOI] [PubMed] [Google Scholar]
- 2.Robitzek E H, Selikoff I J. Am Rev Tuberc. 1952;65:402–428. doi: 10.1164/art.1952.65.4.402. [DOI] [PubMed] [Google Scholar]
- 3.Youatt J. Am Rev Respir Dis. 1969;99:729–749. doi: 10.1164/arrd.1969.99.5.729. [DOI] [PubMed] [Google Scholar]
- 4.Winder F G. In: The Biology of the Mycobacteria. Ratledge C, Stanford J S, editors. London: Academic; 1982. pp. 354–438. [Google Scholar]
- 5.Zhang Y, Young D B. Trends Microbiol. 1993;1:109–113. doi: 10.1016/0966-842x(93)90117-a. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um K S, Wilson T, Collins D, De Lisle G, Jacobs W R. Science. 1994;263:227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 7.Dessen A, Quemard A, Blanchard J S, Jacobs W R, Sacchettini J C. Science. 1995;267:1638–1641. doi: 10.1126/science.7886450. [DOI] [PubMed] [Google Scholar]
- 8.Middlebrook G. Am Rev Tuberc. 1954;69:471–472. doi: 10.1164/art.1954.69.3.471. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Heym B, Allen B, Young D, Cole S T. Nature (London) 1992;358:591–593. doi: 10.1038/358591a0. [DOI] [PubMed] [Google Scholar]
- 10.Musser J M. Clin Microbiol Rev. 1995;8:496–514. doi: 10.1128/cmr.8.4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shoeb H A, Bowman B U, Ottolenghi A C, Merola A J. Antimicrob Agents Chemother. 1985;27:399–403. doi: 10.1128/aac.27.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnsson K, King D S, Schultz P G. J Am Chem Soc. 1995;117:5009–5010. [Google Scholar]
- 13.Kruger-Thiemer E. Am Rev Tuberc. 1958;77:364–367. doi: 10.1164/artpd.1958.77.2.364. [DOI] [PubMed] [Google Scholar]
- 14.Winder F G. Am Rev Respir Dis. 1960;81:68–78. doi: 10.1164/arrd.1960.81.1P1.68. [DOI] [PubMed] [Google Scholar]
- 15.Shoeb H A, Bowman B U, Ottolenghi A C, Merola A J. Antimicrob Agents Chemother. 1985;27:408–412. doi: 10.1128/aac.27.3.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shoeb H A, Bowman B U, Ottolenghi A C, Merola A J. Antimicrob Agents Chemother. 1985;27:404–407. doi: 10.1128/aac.27.3.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deretic V, Philipp W, Dhandayuthapani S, Mudd M H, Curcic R, Garbe T, Heym B, Via L E, Cole S T. Mol Microbiol. 1995;17:889–900. doi: 10.1111/j.1365-2958.1995.mmi_17050889.x. [DOI] [PubMed] [Google Scholar]
- 18.Dhandayuthapani S, Zhang Y, Mudd M H, Deretic V. J Bacteriol. 1996;178:3641–3649. doi: 10.1128/jb.178.12.3641-3649.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sherman D R, Sabo P J, Hickey M J, Arain T M, Mahairas G G, Yuan Y, Barry C E, Stover C K. Proc Natl Acad Sci USA. 1995;92:6625–6629. doi: 10.1073/pnas.92.14.6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christman M F, Morgan R W, Jacobson F S, Ames B N. Cell. 1985;41:753–762. doi: 10.1016/s0092-8674(85)80056-8. [DOI] [PubMed] [Google Scholar]
- 21.Toledano M B, Kullik I, Trinh F, Baird P T, Schneider T D, Storz G. Cell. 1994;78:897–909. doi: 10.1016/s0092-8674(94)90702-1. [DOI] [PubMed] [Google Scholar]
- 22.Wilson T M, Collins D M. Mol Microbiol. 1996;19:1025–1034. doi: 10.1046/j.1365-2958.1996.449980.x. [DOI] [PubMed] [Google Scholar]
- 23.Tai S S, Zhu Y Y. J Bacteriol. 1995;177:3512–3517. doi: 10.1128/jb.177.12.3512-3517.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherman D R, Mdluli K, Hickey M J, Arain T M, Morris S L, Barry C E, Stover C K. Science. 1996;272:1641–1643. doi: 10.1126/science.272.5268.1641. [DOI] [PubMed] [Google Scholar]
- 25.Snapper S B, Melton R E, Mustafa S, Kieser T, Jacobs W R. Mol Microbiol. 1990;4:1911–1919. doi: 10.1111/j.1365-2958.1990.tb02040.x. [DOI] [PubMed] [Google Scholar]
- 26.Jacobs W R, Kalpana G V, Cirrilo J D, Pascopella L, Snapper S B, Udani R A, Jones W, Barletta R G, Bloom B R. Methods Enzymol. 1991;204:537–555. doi: 10.1016/0076-6879(91)04027-l. [DOI] [PubMed] [Google Scholar]
- 27.Beers R F, Sizer I W. J Biol Chem. 1952;195:133–140. [PubMed] [Google Scholar]
- 28.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current Protocols in Molecular Biology. New York: Wiley; 1989. [Google Scholar]
- 29.Rosner J L. Antimicrob Agents Chemother. 1993;37:2251–2253. doi: 10.1128/aac.37.10.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rouse D A, Morris S L. Infect Immun. 1995;63:1427–1433. doi: 10.1128/iai.63.4.1427-1433.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cockerill F R, Uhl J R, Temesgen Z, Zhang Y, Stockman L, Roberts G D, Williams D L, Kline B C. J Infect Dis. 1995;171:240–245. doi: 10.1093/infdis/171.1.240. [DOI] [PubMed] [Google Scholar]
- 32.Heym B, Alzari P M, Honore N, Cole S T. Mol Microbiol. 1995;15:235–245. doi: 10.1111/j.1365-2958.1995.tb02238.x. [DOI] [PubMed] [Google Scholar]
- 33.Morris S L, Bai G H, Suffys P, Portillo-Gomez L, Fairchok M, Rouse D A. J Infect Dis. 1995;171:954–960. doi: 10.1093/infdis/171.4.954. [DOI] [PubMed] [Google Scholar]
- 34.Rouse D A, Li Z, Bai G-H, Morris S L. Antimicrob Agents Chemother. 1995;39:2472–2477. doi: 10.1128/aac.39.11.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pretorius G S, Helden P D, Sirgel F, Eisenach K D, Victor T C. Antimicrob Agents Chemother. 1995;39:2276–2281. doi: 10.1128/aac.39.10.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lirzin M L, Vivien J N, Lepeuple A, Thibier R, Pretet C. Rev Tuberc Pneumol. 1971;35:350–356. [PubMed] [Google Scholar]
- 37.Milano A, De Rossi E, Gusberti L, Heym B, Marone P, Riccardi G. Mol Microbiol. 1996;19:113–123. doi: 10.1046/j.1365-2958.1996.352876.x. [DOI] [PubMed] [Google Scholar]
- 38.Bergler H, Wallner P, Ebeling A, Leitinger B, Fuchsbichler S, Aschauer H, Kollenz G, Hogenauer G, Turnowsky G. J Biol Chem. 1994;269:5493–5496. [PubMed] [Google Scholar]